
Professor Sara Brucker | Professor Olaf Riess | Professor Oliver Kohlbacher – Mayer-Rokitansky-Küster-Hauser Syndrome: Interrogation of Genetic Pathology and Novel Surgical Intervention Methods
{kind=link}
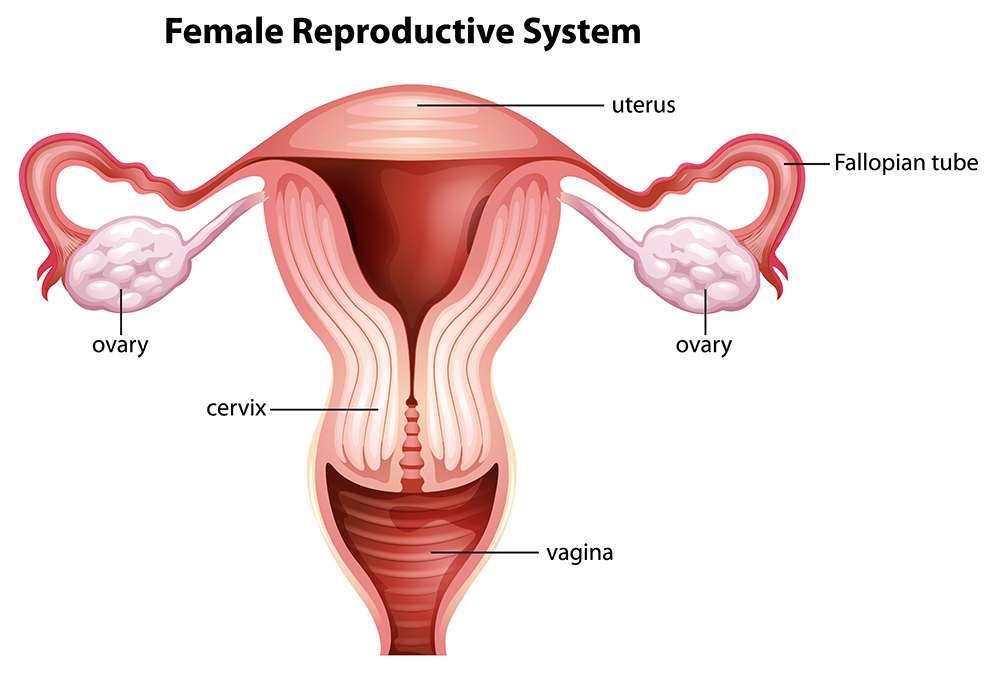
In otherwise phenotypical normal females, that is, females with normal ovaries and regular hormone production, Type I Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome results in the absence of the uterus (womb) and the upper two-thirds of the vagina. Type II MRKH syndrome presents with the same utero-vaginal malformation but also further difficulties that can include structural issues within the urological and skeletal systems. Professors Sara Brucker, Olaf Riess, and Oliver Kohlbacher from the University of Tuebingen are elucidating the molecular pathology of the disorder and have developed a surgical intervention to overcome the major life-changing aspects of this condition.
Mayer-Rokitansky-Küster-Hauser (MRKH): Chance or Genetics?
MRKH syndrome is observed in around one out of every 4,500 live female births. Early research into this rare condition concluded that it is a result of a sporadic defect. However, family clusters in which more family members are diagnosed than would typically be expected by chance have also been observed, indicating a possible genetic basis.
Scientists have investigated many genes in search of the cause of MRKH. However, to date, no single factor has been clearly identified and linked with MRKH syndrome. Professors Sara Brucker, Olaf Riess, and Oliver Kohlbacher from the University of Tuebingen propose that the differences in the disease state of each individual may arise from epigenetic sources, which are the observed genetic discrepancies that have occurred as a result of non-genetic influences.
In one study, Professor Brucker and her colleagues noted that the whole genome and methylation comparisons that they conducted on eight pairs of twins (MRKH/normal) showed a deficiency in both oestrogen receptor and specific HOXA genes, the latter of which are the ‘master regulators’ of embryonic development. This led the team to propose a putative role for either one or both of these in deficiencies in the abnormal development associated with MRKH syndrome.
In addition, in deciphering the pathology of this condition, Professor Brucker and colleagues suggested a putative role for the anti-mullerian hormone promoter, the activation of which may lead to the regression of the Mullerian ducts that typically results in the formation of the uterus and top two-thirds of the vagina during the development of the embryo.

Genetic Mosaicism: Occurrence of Genetic Discrepancies in MRKH Syndrome Females
Continuing their work on the comparison of monozygotic twins, where one twin has MRKH and the other without the condition is the control, the researchers used tissue, blood, and saliva samples to identify two genes which have known functionality in embryonic development of the uterus and endometrium. Testing the tissues and saliva of the MRKH syndrome individuals showed that the genetic errors were present in the uterine tissue but not in the saliva. Such cases, in which a genetic change is observed only in some tissues and not throughout all the tissues in the body, is known as tissue-specific genetic mosaicism. Somatic mutations, that is, mutations that occur in cells other than sperm and egg (germ cells), resulting in mosaicism may explain the reason why we observe cases of monozygotic twins and explain why the genetic children of MRKH syndrome mothers, via surrogacy, do not have the condition.
Genetic Variants of OXTR and ESRI Genes: Their Link with MRKH
Focusing on two genes that have been demonstrated to have a likely effect in MRKH syndrome, the oxytocin receptor gene (OXTR) and the oestrogen receptor I gene (ESRI), Professor Brucker and colleagues sequenced genes from over 90 patients in an attempt to identify the key variants that are linked to the altered expression of these receptors in MRKH syndrome, specifically, lower oxytocin receptor levels and higher oestrogen receptor levels. In doing so, they identified three variants of OXTR and six variants of ESRI that are likely to be associated with MRKH syndrome.
These findings contribute to the hypothesis that hormone receptor deficiency may be a contributory cause of this syndrome. Professor Brucker and colleagues, however, do recognise that their study was limited due to the small number of participants, and they point to the need for larger studies to support their findings.
Comparison of methods for Neovaginal Creation
In addition to the focus on elucidating the genetic pathology of MRKH syndrome, Professor Brucker has successfully developed a surgical innovation to create a vagina in female sufferers that enables them to participate in vaginal intercourse. They conducted an initial study to compare the standard method of neovaginal construction with their new and innovative approach. This confirmed that the new method resulted in shorter operative and traction times, a longer neovagina with improved functional results, fewer surgical complications, and no associated technical issues with the procedure.
Professor Brucker and colleagues recommend this method, which is based on stretching the vaginal dimple, the existing part of the vagina in MRKH syndrome females, with a modified traction device which is applied via laparoscopic surgery.
A follow-up study, examining the longer-term outcomes of 240 patients, demonstrated that this innovative methodology results in long term anatomical stability, with all patients noting that the size of the neovaginas remained stable at 11+ months post-operatively. Importantly, even patients who did not wear the vaginal dummy or who did not achieve sexual intercourse retained anatomical stability. Furthermore, over 70% of the patients, a figure comparable with that of the general population, reported long term functional success, measured as an ability to achieve successful, satisfactory coitus.
Investigating the potential negative effects of the treatment in the longer term, the team assessed multiple parameters, including vaginal prolapse, malignancy, and risk of human papillomavirus infection. The researchers note that patients who failed to follow the necessary protocol, specifically with regards to wearing the vaginal dummy exactly as prescribed, showed some failure, with excessive use of the dummy causing tissue granulation. Meanwhile, failure to use the dummy post-operatively at all resulted, in some cases, in decreased length of the neovagina.
Professor Brucker and colleagues note, that in comparison to non-surgical interventions, such as the use of vaginal dilators, the surgical method produced much more stable results in a more timely fashion. Compared to other surgical methods, like using bowl or skin for a neovagina, there is until now no vaginal prolapse or malignancy or scarring occurred with their method in a very long-term follow-up. The team also highlight the important psychological benefits to the patient of their improved surgical approach.

Surgical Intervention to Enable Pregnancy in MRKH Syndrome Females
Following on from their innovative work on developing a more robust, less problematic neovaginoplasty methodology, Professor Brucker and colleagues have recently presented a review of the processes required to enable an MRKH syndrome female to carry her own biological child. In addition to neovaginoplasty, this also requires uterine transplantation.
Infertility is considered the single most important factor in MRKH syndrome for many women, and with surrogacy illegal in some European and Nordic countries, and many other countries globally, the only option available for these women previously was adoption.
Uterine transplantation can occur from either a live or deceased donor, although a live donation is preferable as pre-operative assessment can ensure suitability and coordination of operations may also be scheduled effectively. Unfortunately, live donor donation carries risks for the donor, including post-operative issues which raise ethical concerns. Furthermore, there may be a psychological risk to the donor if the recipient fails to achieve a pregnancy and live birth following transplantation.
Following extensive animal testing, the first uterine transplants in Sweden showed high success rates, with seven out of the nine patients achieving one or more pregnancies and live birth. Of these seven women, six were MRKH syndrome patients and the seventh had previously undergone surgical removal of her uterus due to cervical cancer. Professor Brucker and her colleagues stressed that there is a substantial amount of monitoring required after the transplantation to ensure the functionality of the transplanted organ is suitable to maintain a pregnancy and live delivery.
Excitingly, by 2019, the collaboration between the Swedish and German scientists had already produced 11 live births from 18 uterine transplants, with the team in Germany having already transplanted four uterus and two live-births. Globally, a team of researchers in Brazil have succeeded in achieving a live birth following a deceased donation, and there is a total of three reported live births that the team is aware of.
Looking to the Future
While Professor Brucker’s collaborations on uterine transplantation are on-going and in their early stages, she is still maintaining a focus on investigating the genetic pathology of the disease. Multiple genes are involved in the early stages of urogenital development, and it may be the case that many genes are involved within the same pathway, but at differing levels, which may partially explain the array of levels of severity observed in MRKH syndrome. To untangle this complex scenario, Professor Brucker and colleagues now propose to generate and analyse cases from discordant monozygotic twins, familial cases, and family trios (parents and child) in spontaneous cases of MRKH.
Reference
https://doi.org/10.33548/SCIENTIA502
Meet the researchers

Professsor Dr Sara Brucker
Executive Director: Department of Women’s Health
Chair: Research Centre for Women’s Health
University of Tuebingen
Germany
Professor Dr Sara Brucker works as the Director of the Department of Women’s Health and Professor at the University of Tuebingen. Following the completion of her thesis in 1999, she undertook a fellowship in Internal Medicine at the University of Ontario, Canada, and thereafter carried out subsequent residencies in Gynaecology and Obstetrics at her current University. A board certified gynaecologist, Professor Brucker is collaborating on an innovative uterine transplantation project that is designed to enable women suffering from Mayer Rokitansky Küster Hauser (MRKH) syndrome which occurs during development of the embryo that results in the absence of a uterus. Professor Brucker’s interests extend to investigating the genetic basis of this condition.
CONTACT
E: sara.brucker@med.uni-tuebingen.de
W: https://www.medizin.uni-tuebingen.de/de/das-klinikum/mitarbeiter/profil/1809

Professor Dr Oliver Kohlbacher
Department of Computer Science
Eberhard Karls University
Tübingen
Germany
Professor Dr Oliver Kohlbacher completed his PhD (2001) in Computer Science at Saarland University/Max Planck Institute for Informatics, Saarbrücken. He is currently a Professor of Applied Bioinformatics and the Director of the Interfaculty Institute for Biomedical Informatics at the University of Tübingen, and Director of the Institute for Translational Bioinformatics at the University Hospital Tübingen. Professor Kohlbacher’s team contribute to the management and analysis of the large quantities of data produced in the assessment of the genetic pathology of MRKH syndrome, which aligns with his interest in personalised medicine and the analysis of biological high-throughput data.
CONTACT
E: oliver.kohlbacher@uni-tuebingen.de

Professor Dr Olaf Riess
Professor of Human Genetics
Director: Institute of Human Genetics and Applied Genomics
University of Tübingen
Germany
Professor Dr Olaf Riess is a Professor of Human Genetics and Director of the Institute of Human Genetics and Applied Genomics at the University of Tübingen. Since completing his MD in 1990, Professor Riess has had a highly successful research career, leading innovation in the university across a number of departments. In 2014, he was a founding member of the Centre of Personalised Medicine. Professor Riess’s work on human genetics and genomics is pivotal in determining the genetic basis of the pathology of MRKH syndrome, and in generating the complex genetic data necessary to investigate the multiple scenarios of its presentation.
CONTACT
E: olaf.riess@med.uni-tuebingen.de
W: https://rd-neuromics.eu/contact/olaf-riess/

Creative Commons Licence
(CC BY 4.0)
This work is licensed under a Creative Commons Attribution 4.0 International License. 
What does this mean?
Share: You can copy and redistribute the material in any medium or format
Adapt: You can change, and build upon the material for any purpose, even commercially.
Credit: You must give appropriate credit, provide a link to the license, and indicate if changes were made.
More articles you may like

Dr Rhett Martin | Governing Nature at the Edge of Collapse
For millions of men, ageing brings with it a set of frustrating and often disruptive urinary symptoms. These symptoms, caused by benign prostatic hyperplasia, or BPH, can affect sleep, confidence, and overall quality of life. Traditionally, treatment follows a familiar path. Patients begin with medications, often for years, and may eventually progress to surgery if symptoms worsen. Yet this pathway is not without its drawbacks. Medications can cause side effects, while surgery carries risks and recovery time. In recent years, a minimally invasive interventional radiology procedure called prostate artery embolisation, or PAE, has begun to challenge this traditional model. At the forefront of this shift is a collaborative research group, led by Dr. Nicholas Brown of the University of Queensland, whose series of P-EASY studies has explored whether PAE could transform how BPH is treated, particularly at earlier stages.

Assoc Prof. Nicholas Brown | Rethinking Prostate Care: A New Frontier in Treating Benign Prostatic Hyperplasia
For millions of men, ageing brings with it a set of frustrating and often disruptive urinary symptoms. These symptoms, caused by benign prostatic hyperplasia, or BPH, can affect sleep, confidence, and overall quality of life. Traditionally, treatment follows a familiar path. Patients begin with medications, often for years, and may eventually progress to surgery if symptoms worsen. Yet this pathway is not without its drawbacks. Medications can cause side effects, while surgery carries risks and recovery time. In recent years, a minimally invasive interventional radiology procedure called prostate artery embolisation, or PAE, has begun to challenge this traditional model. At the forefront of this shift is a collaborative research group, led by Dr. Nicholas Brown of the University of Queensland, whose series of P-EASY studies has explored whether PAE could transform how BPH is treated, particularly at earlier stages.

Jean Lycke | Addressing Unmet Medical Needs in Mucosal Disease: A Close-to-Market Innovation Approach
Recurrent Aphthous Stomatitis (RAS) is an oral condition characterized by one or several painful mucosal ulcers. RAS affects a large proportion of the population and has a point prevalence of approximately 2–3%, daily. The etiology remains unknown, and there is currently no curative treatment. Most patients experience recurring episodes over time, with each episode typically lasting up to a week. Here, we describe the development of a mucoadhesive patch which, when applied over a RAS ulcer, provides rapid pain relief. The patch is easy for patients to apply when symptoms begin and has the potential to be used as an over-the-counter product. The development of the Mucocort mucoadhesive patch is an example of a Close-to-Market innovation strategy that embraces simplicity within a complex healthcare system. By simplifying the product concept, the team has reduced the number of regulatory steps required before market approval. This MedTech/Pharma innovation model, known as the “4R” framework – Re-purposing, Re-formulation, Re-positioning, and Re-patenting – has guided the program from concept to commercialization. In addition to the biodegradable mucoadhesive patch developed for RAS ulcers, the team is extending the innovation concept to a mucoadhesive gel formulation for the prevention and treatment of chemotherapy-induced mucositis. This gel-based program is being commercialized separately through MucoShield.
The Translational Asian Agerelated Macular Degeneration Program Phase 2 (TAAP-2): Reimagining the Future of Vision Care
Age-related macular degeneration, often abbreviated as AMD, is one of the leading causes of vision loss among older adults worldwide. In Asia, where populations are ageing rapidly, its impact is particularly profound. For many, the disease quietly erodes central vision, making everyday activities such as reading, driving, and recognising faces increasingly difficult. Against this backdrop, the Translational Asian Age-related Macular Degeneration Programme, or TAAP for short, has emerged as a bold and ambitious effort to confront the disease headon. Now in its second phase, TAAP-2 represents a significant evolution in both scientific scope and clinical ambition.