
Dr James E. Goldman | Dr Osama Al-Dalahmah – Confronting the Challenge of Huntington Disease
{kind=link}
Huntington disease (HD) is an inherited and progressive neurological disorder which is currently fatal. Dr James E. Goldman and Dr Osama Al-Dalahmah, both at Columbia University, USA, are utilising new techniques in molecular biology to better understand the brain pathology associated with HD. Their vision is to develop therapeutics that can slow the progression of the disease, and ultimately, treat and even prevent it.
A Neurodegenerative Disease Without Cure
Huntington disease (HD) causes severe and wide-ranging neurological problems. It has a prevalence of between 10 and 14 individuals per every 100,00 in Western populations but much lower rates of prevalence are found in Asian and other non-European groups. Diagnosis of HD is based on a confirmed family history or a positive genetic test along with disturbance to motor function (that is, physical movement).
In the early stages, patients may report emotional difficulties such as depression and anxiety, and cognitive difficulties such as trouble learning new information or making decisions. These difficulties increase with disease progression, along with personality changes. Motor impairments, which typically start as small involuntary or twitching movements, known as chorea (from the ancient Greek word ‘choreia,’ meaning dancing) also worsen. Thus, as the diseases progresses, patients may experience difficulties with walking, speaking and eventually swallowing, all severely impacting on their physical and psychosocial wellbeing.
Symptoms usually appear in adults aged in their 30s and 40s, and being a neurodegenerative disease that is without a cure, HD is unreservedly fatal. Life expectancy is usually only around 15 years from the first onset of symptoms. In the advanced stages of the disease, patients require total support in all their daily activities. The final cause of death is usually a complication secondary to HD, such as pneumonia, heart failure or infection.
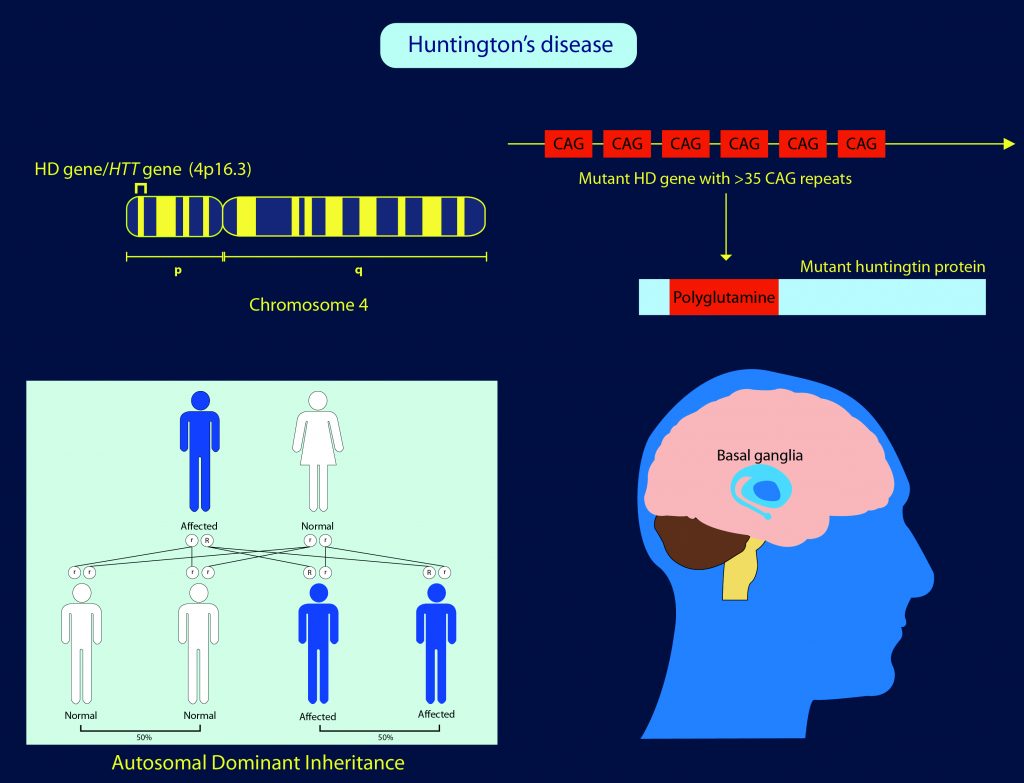
The Genetic Basis of Huntington Disease
HD is an inherited disease, meaning that it is passed down through our deoxyribonucleic acid (DNA), the hereditary material of humans and almost all other organisms. Inside almost all the cells of the body, DNA is contained in a specific part of a cell called the nucleus. Genetic information is stored in DNA in functional units called genes. In the cell, information is transferred from the nucleus (DNA) to the cell machinery that makes the proteins. To do this, the information is copied into ribonucleic acid (RNA) inside the nucleus, and then the RNA is transported outside of the nucleus, where it instructs the cell machinery to make proteins.
A fault in the DNA (or gene) is known as a ‘mutation’ because the protein then encoded by the DNA will be defective and this can result in the development of serious disease, including HD. While many genetic diseases are not passed on to children unless both parents carry the mutation, HD has an autosomal dominant pattern, meaning a child only has to inherit the mutated gene from one parent to develop the disease.
The mutation for HD is found in the HTT gene (also known as the HD gene), which is responsible for making a protein known as huntingtin, which appears particularly important for nerve cells (neurons) in the brain. Even though all of the cells in HD patients carry the HD mutation, the only cells affected appear to be those in the central nervous system, consistent with the neurological effects of the disease. However, within the brain, there is substantial variation in the extent of pathology across different regions. Obtaining a better understanding of why some regions of the brain are more vulnerable, and others more resilient to HD pathology is critical to the development of new therapies for the disease. Exploring this important question is the focus of Dr James E. Goldman and Dr Osama Al-Dalahmah at Columbia University, USA.
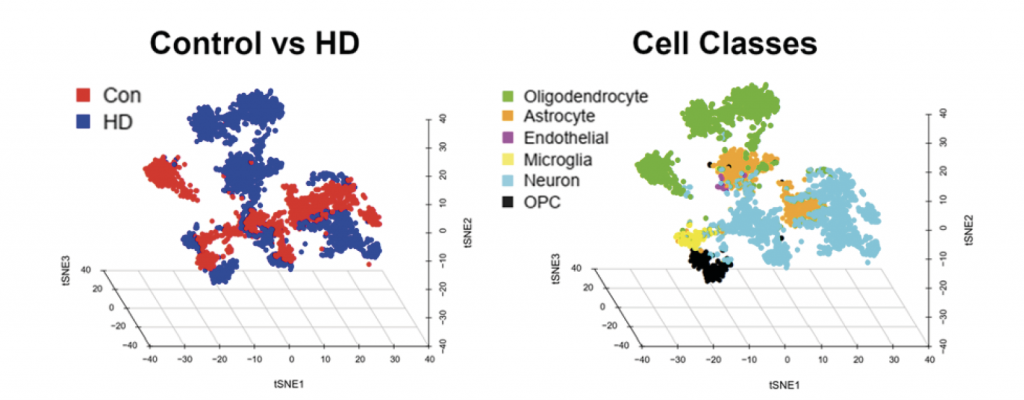
In the figure above, each dot represents a single cell nucleus. The researchers used the specific sets of RNAs to tell them what types of cells each nucleus belongs to. Note that the dots are clustered so that nuclei of similar cell types, defined by their RNAs, are arranged together. On the right, we display the nuclei corresponding to the different cell types in the brains of Huntington (HD) and non-neurological disease patients, all artificially colour coded. Oligodendrocyte precursor cells are an immature cell type present in the adult brain. On the left, the same set of nuclei are displayed, but here all nuclei from the HD brains are in blue and those from the non-neurological patients’ brains in red. Note that nuclei separate into disease vs. non-disease. This means that HD cells produce significant differences in the RNAs from those in non-neurological (CON) brain cells. Understanding these differences is giving us insights into the underlying molecular mechanisms that produce HD and will suggest therapeutic approaches.Reproduced with permission from Al-Dalahmah et al, Single-nucleus RNA-seq identifies Huntington disease astrocyte states, Acta Neuropathologica Communications, 2020.
Harnessing New Methodologies to Study Brain Pathology
Along with colleagues, Dr Goldman and Dr Al-Dalahmah have very recently used a novel, powerful methodology known as single nucleus RNA sequencing (snRNASeq) to explore how RNA changes in the brains of patients with HD compared to healthy individuals. As this cannot be conducted in patients while they are alive, Dr Goldman and the team used specimens from the New York Brain Bank at Columbia University Medical Center, following patient post-mortem and the removal of the brain for freezing and preservation for research.
The cingulate cortex within the brain is associated with functions ranging from cognition, emotion and behaviour, and has extensive connections to other brain structures. It is often affected in HD, and as such, was the region of particular interest to Dr Goldman and Dr Al Dalahmah. They studied patients with HD at grade III/IV level of disease progression, representing the late intermediate stage and the advanced stage, respectively.
Using snRNASeq, the researchers were able to extract intact nuclei from all of the cells in the frozen brain tissues of patients with HD and healthy individuals. As described above, these nuclei contain RNA that has been copied from the cell’s DNA. The RNA was isolated from the nuclei of the individual cells and it was then determined which RNAs were present in each cell. Specifically, this allowed the researchers to identify what genes in the DNA had been copied into the RNAs.
Importantly, not every cell in the human body makes RNAs from every gene, and many genes are, in fact, not copied into RNA across all of the cell types. As Dr Goldman explains, this difference is what makes a cell in the liver different from a cell in the heart, for example. Similarly, there are different cell types in the brain, and each of these copies different genes into RNA. Therefore, it is possible to determine which genes are made into RNAs in the different brain cell types. By comparing these RNAs to those extracted from nuclei of the healthy control individuals, Dr Goldman, Dr Al-Dalahmah and colleagues could see how the RNAs in HD differ as a result of the disease.
They found many changes in the RNAs in HD in all brain cell types, including nerve cells, glial cells, and cells of the immune system. Glial cells are abundant in the brain and two common types are astrocytes and oligodendrocytes. Astrocytes are important in providing critical support for all neurons, while oligodendrocytes make myelin sheaths which wrap around long nerve cell processes called axons, to insulate them and allow very rapid conduction of information.
They found that in HD, while many of the astrocytes appear to try to protect neurons, others appear to be toxic to neurons. The oligodendrocytes are also changed, such that they fail to make myelin properly, meaning that they interfere with the functions of neurons. These findings represent an important step forward in understanding single cell gene expression in HD, and provide a fascinating insight into the role of glial cells, in particular, in HD brain pathology.
‘Our studies will allow us to formulate therapies to slow down the progression of Huntington disease.’
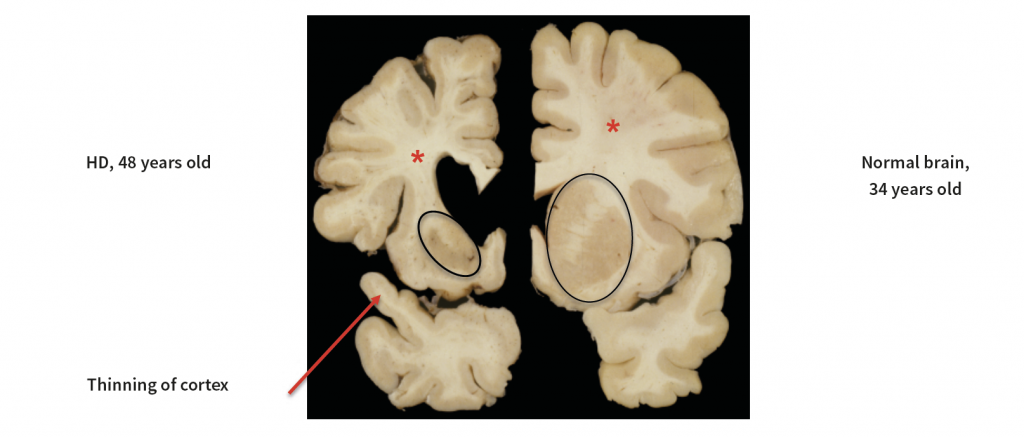
This figure shows a slice of one half of a brain from a Huntington disease (HD) patient on the left, and a comparison with a patient without the disease on the right. The HD brain shows a loss of tissue in a region called the basal ganglia (black ovals) and a thinning of the cerebral cortex (one area of cortex is shown by arrow). Note also the loss of white matter (*), which contains the long axon processes by which neurons communicate with other neurons in the brain. Courtesy of Dr Jean Paul Vonsattel, Columbia University Irving Medical Center
Future Work and a Therapeutic Vision
Building on this work, the investigators are now comparing RNAs from patients in different stages of severity of HD and different regions of the HD brain. In particular, they will look for differences in the brains of individuals who have died in the early stages of HD from those in the late stages of the disease. This is critical, given that HD is a long-term, progressive disease.
Targetting RNA and DNA from a therapeutic perspective is described in the literature as the most promising approach we currently have in the efforts to confront and meet the challenge of HD. Dr Goldman reveals his vision that ‘Our studies will allow us to formulate therapies to slow down the progression of Huntington disease.’ Dr Goldman and colleagues are also committed to sharing their data as a resource for all researchers in the field of HD, further driving forward the ultimate goal of treating and, one day, even preventing this currently fatal disease.
Dr Goldman and Dr Al Dalahmah soon will begin to study the brains of patients with Parkinson disease in the same way, isolating cell nuclei and determining what RNAs are expressed in the variety of cell types. They hope to discover the changes that accompany the development of Parkinson disease, a common neurodegenerative disorder that results in tremors, slowness of movement and in some patients, a loss of their cognitive abilities. They anticipate that some of the changes in the Parkinson brain will mirror those in HD, while there may be changes specific to each disease. With other investigators who are pursuing similar strategies in other neurological diseases, they eventually will contribute to an understanding of these diseases at a new and deep molecular level.
Reference
https://doi.org/10.33548/SCIENTIA566
Meet the researchers

Dr James E. Goldman
Professor of Pathology and Cell Biology
Columbia University College of Physicians & Surgeons
New York, NY
USA
Dr James E. Goldman completed his MD and his PhD in neurobiology at New York University School of Medicine in 1976. In 1987, he moved to the Columbia University College of Physicians & Surgeons, where he is now Professor of Pathology and Cell Biology. As a practising neuropathologist for over 30 years, Dr Goldman has a strong background in central nervous system pathology and broad expertise in the cellular and molecular pathology of neurological diseases. His current work includes the use of fresh frozen human brain tissue from the New York Brain Bank at Columbia University Medical Center to investigate gene expression of individual cell types in Huntington disease and other neurological disorders. Dr Goldman has published prolifically in prestigious journals over his career, which serves as testimony to his outstanding contribution to science.
CONTACT
E: jeg5@cumc.columbia.edu

Dr Osama Al-Dalahmah
Columbia University Medical Center
New York, NY
USA
Dr Osama Al-Dalahmah completed an MD in medicine at the University of Jordan in 2010 and then a PhD in developmental neuroscience at the University of Oxford, UK, in 2015. Following postdoctoral research at the same institution, he moved to Columbia University, USA, in 2016, where he is currently an Instructor in Neuropathology. With nearly a decade of clinical and research experience in neurobiology, Dr Al-Dalahmah is supported by ongoing funding and maintains an active publication record in his scientific field. Notably, he is the first recipient of the Nancy S. Wexler Discovery Fund Young Investigator Prize from the Hereditary Disease Foundation for his work on Huntington’s Disease.
CONTACT
E: oa2298@cumc.columbia.edu
KEY COLLABORATORS
Vilas Menon, Department of Neurology, Columbia University
Jean Paul Vonsattel, Department of Pathology & Cell Biology, Columbia University
Alexander Sosunov, Department of Neurosurgery, Columbia University
Istvan Adorjan, Department of Anatomy, Histology and Embryology, Semmelweiss University, Budapest, Hungary
Abdullah Ahmed Shaik, College of Medicine, Alfaisal University, Riyadh, Saudi Arabia
Kenneth Ofori, Department of Pathology & Cell Biology, Columbia University
Yang Liu, Department of Pathology & Cell Biology, Columbia University
FUNDING
Hereditary Disease Foundation
Huntington’s Disease Society of America
Taub Institute for Research on Alzheimer’s Disease and the Aging Brain, Columbia University
Department of Pathology & Cell Biology, Columbia University
![]()
![]()
![]()
![]()
![]()
Want to republish our articles?
We encourage all formats of sharing and republishing of our articles. Whether you want to host on your website, publication or blog, we welcome this. Find out more
Creative Commons Licence
(CC BY 4.0)
This work is licensed under a Creative Commons Attribution 4.0 International License. 
What does this mean?
Share: You can copy and redistribute the material in any medium or format
Adapt: You can change, and build upon the material for any purpose, even commercially.
Credit: You must give appropriate credit, provide a link to the license, and indicate if changes were made.
More articles you may like

Jean Lycke | Addressing Unmet Medical Needs in Mucosal Disease: A Close-to-Market Innovation Approach
Recurrent Aphthous Stomatitis (RAS) is an oral condition characterized by one or several painful mucosal ulcers. RAS affects a large proportion of the population and has a point prevalence of approximately 2–3%, daily. The etiology remains unknown, and there is currently no curative treatment. Most patients experience recurring episodes over time, with each episode typically lasting up to a week. Here, we describe the development of a mucoadhesive patch which, when applied over a RAS ulcer, provides rapid pain relief. The patch is easy for patients to apply when symptoms begin and has the potential to be used as an over-the-counter product. The development of the Mucocort mucoadhesive patch is an example of a Close-to-Market innovation strategy that embraces simplicity within a complex healthcare system. By simplifying the product concept, the team has reduced the number of regulatory steps required before market approval. This MedTech/Pharma innovation model, known as the “4R” framework – Re-purposing, Re-formulation, Re-positioning, and Re-patenting – has guided the program from concept to commercialization. In addition to the biodegradable mucoadhesive patch developed for RAS ulcers, the team is extending the innovation concept to a mucoadhesive gel formulation for the prevention and treatment of chemotherapy-induced mucositis. This gel-based program is being commercialized separately through MucoShield.
The Translational Asian Agerelated Macular Degeneration Program Phase 2 (TAAP-2): Reimagining the Future of Vision Care
Age-related macular degeneration, often abbreviated as AMD, is one of the leading causes of vision loss among older adults worldwide. In Asia, where populations are ageing rapidly, its impact is particularly profound. For many, the disease quietly erodes central vision, making everyday activities such as reading, driving, and recognising faces increasingly difficult. Against this backdrop, the Translational Asian Age-related Macular Degeneration Programme, or TAAP for short, has emerged as a bold and ambitious effort to confront the disease headon. Now in its second phase, TAAP-2 represents a significant evolution in both scientific scope and clinical ambition.

Ms. Aikaterini Dritsoula | Looking Beyond Snoring: How Hidden Airway Problems Shape Children’s Sleep
For many parents, a child’s snoring may seem harmless, even endearing. Yet in some cases, it signals something more serious. Obstructive sleep apnoea is a condition in which a child’s breathing is repeatedly disrupted during sleep. These interruptions can affect growth, behaviour, and learning. Children with this condition may toss and turn at night, struggle to concentrate during the day, or show signs of hyperactivity and fatigue. Traditionally, enlarged tonsils and adenoids have been seen as the main culprits. Surgery to remove them has long been considered the standard treatment. However, research led by Consultant ENT Surgeon Ms. Aikaterini Dritsoula of The Leeds Teaching Hospitals NHS Trust invites us to look deeper. Her work suggests that the story is often more complex, especially in very young children.

Sara F Martin | The New Paradigm: Two Fundamental 22-year Solar Cycles Always Present on the Sun
For millennia, humans have looked up towards the life-giving Sun and sought to understand its nature. One of its earliest features noticeable before the age of technology was the presence of small dark patches scattered across its surface – sunspots. These blemishes appeared to wax and wane on a regular 11-year cycle, which was thought for over a century to be a fundamental time period governing the Sun’s magnetic activity. But new discoveries suggest a radically different understanding where sunspots are merely peak phases of two, more fundamental 22- year magnetic cycles present simultaneously in different bands of latitude.