
Dr Y-H Taguchi – In Silico Drug Discovery for COVID-19 Using an Unsupervised Feature Extraction Method
{kind=link}
In silico drug discovery is useful for screening and identifying large numbers of drug candidate compounds in a way that is not possible using classical experimental approaches. Dr Y-H Taguchi at Chuo University, Japan, has developed a computational technique known as ‘tensor decomposition-based unsupervised feature extraction’. He has successfully applied this as an in silico phenotype-based drug discovery method to repurpose known drugs for severe acute respiratory syndrome coronavirus 2 and has successfully identified various known anti-viral drugs as viable candidates for the successful treatment of COVID-19.
A Mathematical Framework for In Silico Drug Discovery
Since January 2020, the COVID-19 pandemic has critically affected communities worldwide, prompting scientists to identify new, effective drugs that could tackle the disease. To repurpose old drugs toward the treatment of COVID-19, we must first understand the mechanism by which SARS-CoV-2 successfully invades human cells, causing the onset of disease. Driven by advances in information technology, a new approach, known as in silico experimentation, has generated reports of a large number of candidate drug compounds that may be useful for treating COVID-19. In biomedical research, an in silico experiment is one that is conducted with the aid of computer simulations.
Dr Y-H Taguchi and his team from the Department of Physics, Chuo University, Japan, have developed computational techniques that can support in silico experimentation, allowing researchers to predict the function of proteins, discover potential drug-like molecules and identify disease-causing genetic mutations.
Since disease alters gene expression, it is not surprising that there are specific sets of genes for which altered expression patterns can act as biomarkers to identify the presence of disease and estimate disease progression. Dr Taguchi and his collaborators had previously used a mathematical method known as ‘tensor decomposition (TD)-based unsupervised feature extraction (FE)’ and applied it to a gene expression profile dataset obtained from mouse liver infected with the mouse hepatitis virus, regarded by many as a suitable model of human coronavirus infection. The results of the study were recently published in April 2021.

The main purpose of the methods developed by Dr Taguchi is to perform feature selection, which means selecting a small or limited number of critical variables from a very large number of variables. Feature selection strategies can be classified into supervised ones and unsupervised ones. Generally, supervised strategies are more popular than unsupervised ones. This is because the purpose of feature selection is usually clear to the user. Despite this, the use of unsupervised feature selections provides a better choice when class labels for large sets of data are unclear or unavailable.
In September 2020, Dr Taguchi’s team published the results of the successful application of an unsupervised strategy able to predict anti-COVID-19 drug candidate compounds without prior knowledge of effective known compounds. The team analysed the gene expression profiles of multiple lung cancer cell lines infected with SARS-CoV-2, in the presence or the absence of several antiviral drugs. All the gene expression profiles were obtained from a public database.
SBDD can find drug candidate compounds in the absence of structural similarity to known drugs and requires massive computational resources for ‘docking’ simulations between compounds and proteins. Dr Taguchi’s TD-based unsupervised FE approach successfully overcame the limitations associated with SBDD, predicting a set of effective drug candidate compounds that are able to treat COVID-19.

Tensor Decomposition as a Feature Extraction Method
One classic approach used to identify significant variables is to conduct a statistical test. This test would compute the probability that a desired property can appear by chance rather than being associated with a specific feature. For example, if the alteration of a gene, or set of genes, follows the onset of disease, the probability of it happening by chance would be rather small. In scenarios where there are very large numbers of variables and a small number of observations, as in genomic science, this strategy often fails. To perform feature selection in these scenarios, Dr Taguchi has successfully applied a mathematical approach known as tensor decomposition.
Tensors are a feature of linear algebra and are at the top of a hierarchy that includes scalars, vectors and matrices. Scalars are simple numerical values, such as the mass of an object or the price of an item for sale. Vectors are composed of a set of scalars. The elements that make up vectors are represented by adding a suffix to scalars, e.g., xj, where x is the scalar value and j is a suffix that represents a whole number. This means that the value of the vector depends on both x and j.
As vectors are composed of scalars, matrices, X, are composed of x vectors. Any x vector belonging to a matrix will have to suffixes j and i (xij). For example, the ‘j’ component of vectors in a matrix could be an item such as ‘Bread’, ‘Fish’, or ‘Pork’, which can vary in value within certain categories denoted as ‘i’, with i1, for example, being ‘Mass’, i2 being ‘Price’, i3 being ‘Calories’.
As vectors are composed of scalars and matrices are composed of vectors, tensors can be composed of matrices. Suppose we have some samples of foods in two different shops. Now, we can define a tensor, Xijk, that describes the jth feature, attributed to the ith food, in the kth shop.
The technique of tensor decomposition can be applied to a large number of experimental conditions. For example, if gene expression is measured for various tissues taken from different patients, gene expression is better represented, not in a matrix, but as a tensor, where patients vs tissues vs genes, are the parameters that define the tensor.

Ivermectin: A Promising COVID-19 Treatment
TD-based unsupervised FE was applied to the gene expression profiles of multiple lung cancer cell lines infected with SARS-CoV-2. Five cell lines underwent two different treatments: one with SARS-CoV-2 and one with a ‘mock treatment’. There were 30 samples in the end, as each pair cell line/treatment was analysed in triplicate (5 cell lines x 2 treatments x 3 replicates = 30 samples). Since there is currently a lack of known drugs that are effective in treating SARS-CoV-2, a ligand based drug discovery approach would not be useful because it is based on the known structures of compounds. On the other hand, SBDD requires massive computational resources, like supercomputers, whereas Dr Taguchi’s method can be performed with standard computational servers that can be purchased even with reduced budgets.
The researchers identified several candidate compounds that could significantly alter the expression of the 163 genes selected by TD-based unsupervised FE. The 163 selected genes are all responsible for expressing proteins that significantly interact with the proteome of the SARS-CoV virus, which is closely related to SARS-CoV-2. Numerous drugs were successfully identified, especially antiviral drugs, including fluticasone, atorvastatin, gentamicin, among many others. The screening process detected ivermectin as the promising treatment for COVID-19. Ivermectin, which was previously identified as an anti-parasite drug, was recently included in clinical trials for SARS-CoV-2.

Summing up: Remarkable Progress
Dr Taguchi and his collaborators proposed an advanced unsupervised machine learning method for identifying numerous promising drug candidate compounds that could treat COVID-19 infection. When applied to the expression profiles of a pool of genes from lung cancer cell lines infected by SARS-CoV-2, the method identified numerous drug compounds that significantly altered the expression of the genes, indicating a change in the progression of the disease. The study was aimed at consolidating a similar strategy previously employed by Dr Taguchi to understand the infectious process of mouse hepatitis virus, a well-studied model for COVID-19.
In order to confirm the significance of the 163 genes in the context of human disease, Dr Taguchi and his collaborators compared the genes with those identified to be interacting with SARS-CoV-2 in humans. The 163 genes identified in this study turned out to be associated with human genes previously reported to interact with the SARS-CoV-2 proteome, contributing to disease progression.
Although ivermectin was recently reported to inhibit the replication of SARS-CoV-2 in vitro, to Dr Taguchi’s knowledge, his team was the first to report the in silico detection of ivermectin as a possible SARS-CoV-2 drug through an unsupervised feature extraction method. Most in silico drug discovery methods are supervised strategies that require known target-drug relations or drug-disease relations, which are currently not available for SARS-CoV-2. Furthermore, as ivermectin was first identified as an anti-parasite drug, no previous supervised in silico approach considered it, confirming the remarkable effectiveness of the unsupervised approach devised by Dr Taguchi and his collaborators.
Reference
https://doi.org/10.33548/SCIENTIA727
Meet the researcher

Dr Y-h. Taguchi
Department of Physics
Chuo University
Tokyo
Japan
Dr Y-h. Taguchi obtained his PhD in the theory of statistical mechanics of spin systems, from the Tokyo Institute of Technology in 1988. In the same year, he started his academic career as Assistant Professor at the Department of Physics at the Tokyo Institute of Technology. In 1997, he joined the Department of Physics at Chuo University, Tokyo, where he became Full Professor in 2006. Dr Taguchi’s most recent research interest revolves around the development of tensor decomposition methods applied to bioinformatics, particularly in relation to proteomics and gene expression patterns. Dr Taguchi has published a monograph and several peer-reviewed publications. As an outstanding scientist in his field, Dr Taguchki has received numerous prestigious honours and awards for his contributions to bioinformatics.
CONTACT
E: tag@granular.com
W: https://researchmap.jp/Yh_Taguchi/
Twitter: @Yh_Taguchi
KEY COLLABORATORS
Dr Turki Turki, King Abdulaziz University, Jeddah
FUNDING
KAKENHI (grant numbers 19H05270, 20H04848 and 20K12067)
Deanship of Scientific Research at King Abdulaziz University, Jeddah (grant number KEP-8-611-38)
FURTHER READING
YH Taguchi, T Turki, Application of Tensor Decomposition to Gene Expression of Infection of Mouse Hepatitis Virus Can Identify Critical Human Genes and Effective Drugs for SARS-CoV-2 Infection, IEEE Journal of Selected Topics in Signal Processing, 2021, 15(3), 746–758.
YH Taguchi, T Turki, A new advanced in silico drug discovery method for novel coronavirus (SARS-CoV-2) with tensor decomposition-based unsupervised feature extraction, PLoS ONE, 2020, 15(9), e0238907.
YH Taguchi, Unsupervised feature extraction applied to bioinformatics: PCA and TD based approach, 2020, Switzerland: Springer International.
![]()
Want to republish our articles?
We encourage all formats of sharing and republishing of our articles. Whether you want to host on your website, publication or blog, we welcome this. Find out more
Creative Commons Licence
(CC BY 4.0)
This work is licensed under a Creative Commons Attribution 4.0 International License. 
What does this mean?
Share: You can copy and redistribute the material in any medium or format
Adapt: You can change, and build upon the material for any purpose, even commercially.
Credit: You must give appropriate credit, provide a link to the license, and indicate if changes were made.
More articles you may like

Jean Lycke | Addressing Unmet Medical Needs in Mucosal Disease: A Close-to-Market Innovation Approach
Recurrent Aphthous Stomatitis (RAS) is an oral condition characterized by one or several painful mucosal ulcers. RAS affects a large proportion of the population and has a point prevalence of approximately 2–3%, daily. The etiology remains unknown, and there is currently no curative treatment. Most patients experience recurring episodes over time, with each episode typically lasting up to a week. Here, we describe the development of a mucoadhesive patch which, when applied over a RAS ulcer, provides rapid pain relief. The patch is easy for patients to apply when symptoms begin and has the potential to be used as an over-the-counter product. The development of the Mucocort mucoadhesive patch is an example of a Close-to-Market innovation strategy that embraces simplicity within a complex healthcare system. By simplifying the product concept, the team has reduced the number of regulatory steps required before market approval. This MedTech/Pharma innovation model, known as the “4R” framework – Re-purposing, Re-formulation, Re-positioning, and Re-patenting – has guided the program from concept to commercialization. In addition to the biodegradable mucoadhesive patch developed for RAS ulcers, the team is extending the innovation concept to a mucoadhesive gel formulation for the prevention and treatment of chemotherapy-induced mucositis. This gel-based program is being commercialized separately through MucoShield.
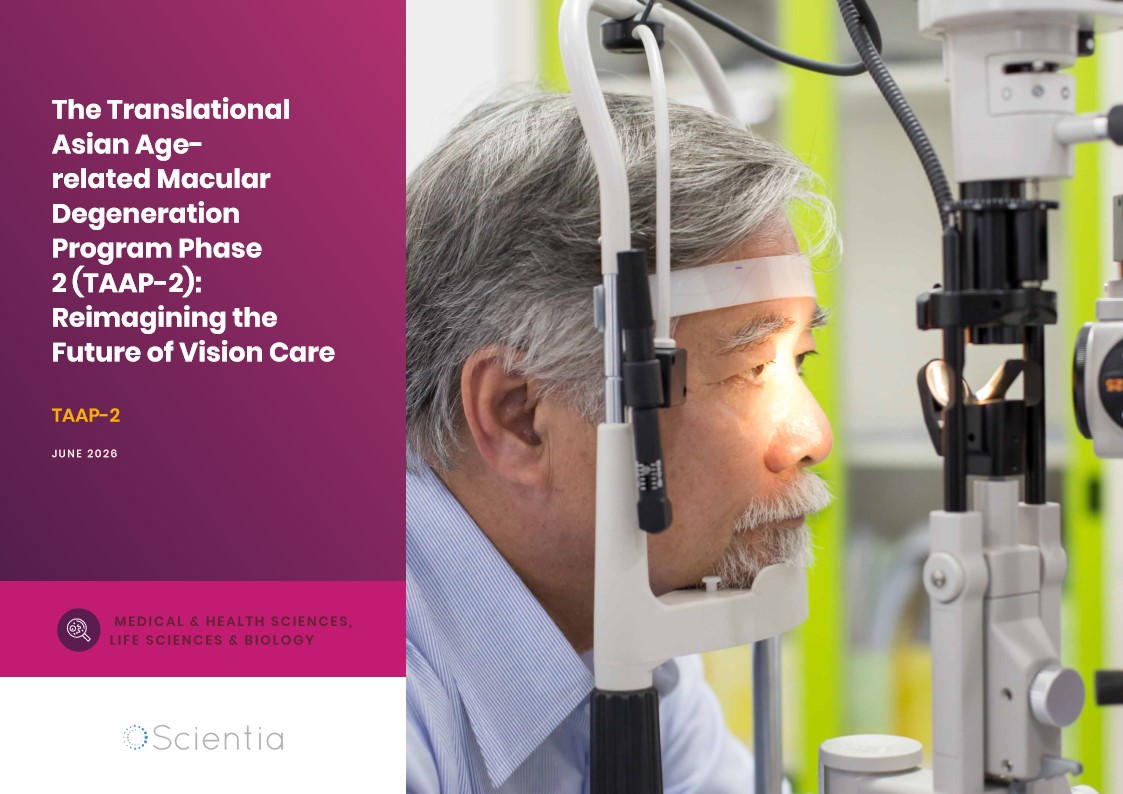
The Translational Asian Agerelated Macular Degeneration Program Phase 2 (TAAP-2): Reimagining the Future of Vision Care
Age-related macular degeneration, often abbreviated as AMD, is one of the leading causes of vision loss among older adults worldwide. In Asia, where populations are ageing rapidly, its impact is particularly profound. For many, the disease quietly erodes central vision, making everyday activities such as reading, driving, and recognising faces increasingly difficult. Against this backdrop, the Translational Asian Age-related Macular Degeneration Programme, or TAAP for short, has emerged as a bold and ambitious effort to confront the disease headon. Now in its second phase, TAAP-2 represents a significant evolution in both scientific scope and clinical ambition.

Ms. Aikaterini Dritsoula | Looking Beyond Snoring: How Hidden Airway Problems Shape Children’s Sleep
For many parents, a child’s snoring may seem harmless, even endearing. Yet in some cases, it signals something more serious. Obstructive sleep apnoea is a condition in which a child’s breathing is repeatedly disrupted during sleep. These interruptions can affect growth, behaviour, and learning. Children with this condition may toss and turn at night, struggle to concentrate during the day, or show signs of hyperactivity and fatigue. Traditionally, enlarged tonsils and adenoids have been seen as the main culprits. Surgery to remove them has long been considered the standard treatment. However, research led by Consultant ENT Surgeon Ms. Aikaterini Dritsoula of The Leeds Teaching Hospitals NHS Trust invites us to look deeper. Her work suggests that the story is often more complex, especially in very young children.

Sara F Martin | The New Paradigm: Two Fundamental 22-year Solar Cycles Always Present on the Sun
For millennia, humans have looked up towards the life-giving Sun and sought to understand its nature. One of its earliest features noticeable before the age of technology was the presence of small dark patches scattered across its surface – sunspots. These blemishes appeared to wax and wane on a regular 11-year cycle, which was thought for over a century to be a fundamental time period governing the Sun’s magnetic activity. But new discoveries suggest a radically different understanding where sunspots are merely peak phases of two, more fundamental 22- year magnetic cycles present simultaneously in different bands of latitude.