
Dr Gábor Csányi – Macropinocytosis: The Macrophage Drinking Problem Behind Atherosclerosis
Dr Gábor Csányi and his team at the Vascular Biology Center of Augusta University explore the molecular mechanisms involved in an alternative pathway of macrophage LDL cholesterol uptake, with hopes of finding new targets for the treatment of atherosclerosis.
HDL, LDL and Atherosclerosis: The Good, the Bad and the Ugly
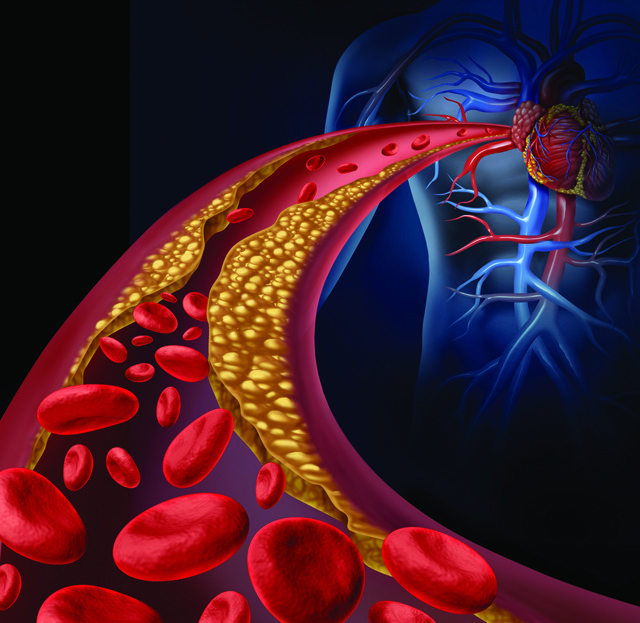
Atherosclerosis is a chronic inflammatory disease in which lipid rich plaques build up in the walls of major arteries, restricting the flow of oxygen-rich blood to different tissues and organs. Atherosclerotic plaques result from the accumulation of fatty deposits along the vessel wall. Growing plaques can protrude into the arterial lumen, eventually causing a partial or complete obstruction of blood flow. Angina pectoris is a medical condition caused by a gradually growing stable plaque and subsequent decrease in coronary blood flow. Unfortunately, atherosclerotic plaques can become unstable and rupture, leading to thrombus formation, rapid decrease in blood flow and low tissue oxygen levels. Atherosclerosis can lead to serious problems, including heart attack, stroke and peripheral artery disease.
The major player in atherosclerosis is cholesterol. As a fat, or lipid, cholesterol cannot be dissolved and float freely in the blood. To overcome this, the body has specific proteins, called lipoproteins, which bind to it and carry it where it needs to go. Two types of cholesterol carriers are low-density lipoproteins (LDLs) and highdensity lipoproteins (HDLs). Clinically, they are known as ‘bad’ and ‘good’ cholesterol, respectively.
Download

Share
{kind=link}
‘These findings support a new paradigm in which redox signaling-mediated lipid macropinocytosis independent of broader lipid oxidation propagates initiation and development of vascular inflammatory disease’
Atherosclerotic lesions typically develop where arteries branch. In response to disturbed blood flow that naturally occurs in these forked regions, blood vessels thicken. When this happens, the endothelial cells lining the interior vascular surface become activated and exhibit increased permeability, making it easier for LDL to enter into the intima – the innermost layer of the vessel wall. Individuals with high cholesterol have abnormally elevated levels of LDL in the blood, which increases the number of LDLs accessing and colonising the vascular wall. A major problem here is that when LDLs are exposed to the new environment, they undergo oxidative modifications that transform them into pro-inflammatory molecules, which release factors from the endothelium that initiate and maintain inflammation in the vessel wall.

Macrophage
Several cells of the immune system respond to these pro-inflammatory signals. One particular type, called monocytes, migrate from the blood to these inflammatory sites in the vessel wall and transform into larger, mature macrophages. Macrophages are armed with the necessary machinery to recognise and remove oxidised LDL from the vascular space. Proteins located along the macrophage membrane, called scavenger receptors, recognise oxidised LDL and bring it inside to be used or eliminated by the cell. However, accumulated LDL in these diseased areas increases the internalisation rate, which transforms these highly mobile removal workers into fat-laden and less mobile ‘foam’ cells. The lipids engulfed by the macrophage give it a ‘foamy’ appearance (red colour on the right). Paralysed, these foam cells die and become part of the expanding fatty plaque they were previously trying to prevent. Over time this plaque expands, narrowing the vessel lumen and reducing the volume of blood that can pass through. If and when this plaque ruptures, a blood clot will emerge and obstruct the vessel entirely, starving the organ it serves of its oxygen supply. If it serves the heart, it can cause a heart attack, while if it occurs in the brain, it is called stroke.
While progress has been made in terms of diagnosis, prevention and treatment, atherosclerosis and its cardiovascular consequences continue to cause high rates of morbidity and mortality worldwide. Therefore, it is important to improve our understanding of the biological mechanisms involved, so that better therapies can be developed.
Pinocytosis: An Unexplored Target in Atherosclerosis
Dr Gábor Csányi and his group have contributed to a better understanding of how macrophages ingest bad cholesterol and transform into lipid-laden foam cells. They have shown that LDLs do not actually need to be oxidised to be internalised, and also, that another mechanism of uptake is involved in macrophage foam cell formation. Essentially, in addition to relying on the selective uptake of modified LDLs by scavenger receptors, macrophages use pinocytosis to ingest unmodified LDLs. Pinocytosis, often referred to as ‘cell drinking’, is the engulfing of extracellular fluid – liquid that surrounds the cell. This process is nonspecific, because the cell swallows everything in the fluid, including random dissolved particles. The rate of pinocytosis and pericellular solute concentration are the primary factors that determine the extent of solute internalisation.
So why do cells risk allowing entry to everything? Immune cells, like macrophages, use pinocytosis to survey

Lipid-laden macrophage or foam cell
the surrounding environment for antigens. They bring samples inside and evaluate them to establish whether there are foreign invaders that might be potentially harmful and need to be eliminated. Additionally, the cells use pinocytosis to internalise extracellular nutrients to support their growth and metabolic needs. Macrophages use two forms of pinocytosis to internalise fluid and associated solutes – macropinocytosis and micropinocytosis. As their names suggest, they ingest large and small volumes of extracellular fluid, respectively. While both types of pinocytosis may participate in the ingestion of unmodified LDLs, Dr Csányi and his group are specifically interested in the role that macropinocytosis plays in this process.
Macropinocytosis starts when extracellular signalling molecules interact with specific protein receptors housed on the cell membrane. When the cell receives the initiating signal, membrane ‘ruffles’ form, creating folds and protrusions that extend into the surrounding environment. These protrusions move back towards the plasma membrane, much like a swimmer doing the breast stroke, capturing fluid and solutes in their wake. Membrane protrusions can also transition into curved ruffles and form ice cone like macropinocytotic cups, which close on the top, capturing extracellular fluid and solutes. Following this, the newly formed fluid sacks pinch off from the plasma membrane to form irregular shaped and sized vesicles called macropinosomes, which travel into the cytoplasm – the cell’s interior.

A ‘thirsty’ macrophage develops membrane ruffles to drink extracellular fluid
Dr Csányi’s research group has delved deeper into the mechanisms by which macropinocytosis of lipids are regulated. ‘My research focuses on a better understanding of the mechanisms leading to atherosclerosis and applying this understanding to develop and implement new strategies for its prevention and treatment,’ he explains. ‘Utilising a broad range of innovative technologies and strategies, we investigate the mechanisms by which cardiovascular risk factors, inflammatory mediators, matrix proteins, and genes promote the transition from a healthy “normal” vessel wall to a “diseased” vasculature.’
Dr Csányi and his team devised several intricate experiments to investigate what molecules and pathways regulate macropinocytosis. The team has identified a new mechanism regulating macropinocytosis of unmodified LDL by macrophages.
The role of Nox2 in Macropinocytosis
In their research, Dr Csányi and his team demonstrated that Nox2 participates in macropinocytosis. Belonging to the NADPH oxidase (Nox) family of protein oxidases, Nox2 functions in generating reactive oxygen species (ROS) and mediating intracellular redox reactions. Briefly, this enzyme complex is responsible for transferring electrons to molecular oxygen to produce superoxide anions – the precursor radicals of ROS. Low levels of ROS regulate signalling pathways that are important for maintaining healthy cellular physiology, while increased ROS generation leads to oxidative stress and contributes to vascular and other diseases.
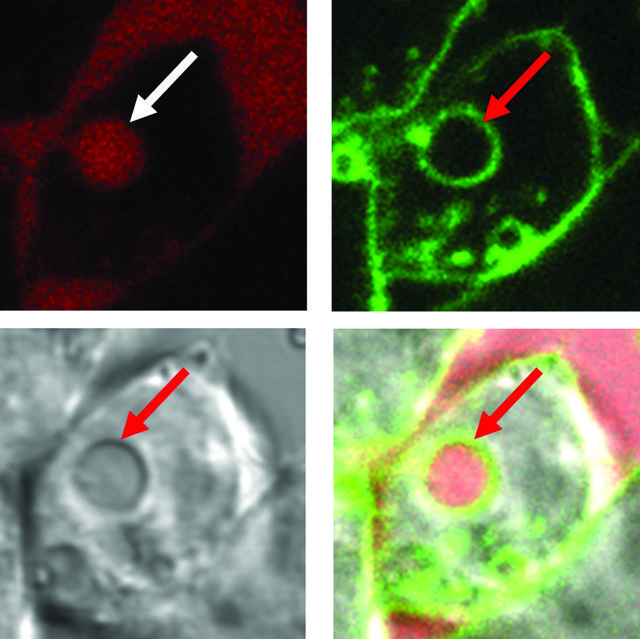
Internalised fluid (arrows)
Dr Csányi chose Nox2 not only because of its importance in ROS production in macrophages, but also because it exists downstream in a signalling chain of molecules, namely Protein Kinase C (PKC) and Ras-1, known to function in macropinocytosis. Therefore, he proposed that these other proteins may send signals to Nox2 to help them initiate macropinocytosis. Dr Csányi and his team conducted their initial experiments using macrophages in the lab. They used 4ß-PMA, a molecule known to induce macropinocytosis in macrophages, to test whether Nox2 participates. A clever way to explore whether a particular molecule is important in a biological process is to disrupt its function. Firstly, they deactivated the Nox2 protein and found that 4ß-PMAinduced macropinocytosis and the amount of lipid ingested was significantly reduced. To clarify these findings, they silenced the Nox2 gene before it was able to be fully translated into functional protein. Again, macropinocytosis was inhibited.
While evaluating the biological processes in cells on their own was informative, it was important to test whether similar results translated to animal models of atherosclerosis. To test this, the team extracted macrophages from wild-type mice, whose genetics have not been manipulated, and Nox2 knockout mice, where Nox2 expression is genetically removed. These macrophages were treated with 4ß-PMA to induce macropinocytosis and subsequently injected into the peritoneal cavities of ApoEˉ/ˉ mice–bred to develop high cholesterol similar to that seen in atherosclerotic patients. A day later, Dr Csányi and his colleagues extracted and analysed the macrophages and found that macropinocytosis and lipid uptake was significantly diminished in those taken from the Nox2 knockout mice. Cumulatively, these experiments highlighted Nox2 as a principal player in the induction of macropinocytosis. Next, Dr Csányi’s laboratory sought to identify the signalling mechanisms downstream of Nox2 involved in macropinocytosis.
In further investigations, Dr Csányi and his team found that intracellular Nox2 signalling activates cofilin (an actin-binding protein) to trigger membrane ruffling – the first stage of macropinocytosis. It does this by stimulating a secondary signalling pathway, called the phosphoinositide 3 kinase/Akt pathway (PI3K/Akt). For the first time, Dr Csányi and his colleagues demonstrated that Nox2-mediated intracellular redox signalling plays a pivotal role in a mechanism that initiates macrophage LDL macropinocytosis.

CD47 and Nox1: Partners in Crime
Dr Csányi, Dr Pagano and their colleagues defined a physiologically relevant pathway that mediates macrophage macropinocytosis of unmodified LDL. The extracellular matrix surrounding a cell’s exterior provides structural and biochemical stability. It is composed of different molecules, including a group of matricellular proteins, which help to regulate pathways that are important for normal cell maintenance. One type of matricellular protein, called thrombospondin-1 (TSP1), is elevated above healthy levels in the intimal layer of the vessel wall in individuals with atherosclerosis. Therefore, Dr Csányi proposed that TSP1 may act as a physiological stimulator of LDL macropinocytosis in macrophages.
The research team found that incubating macrophages with physiologically relevant concentrations of TSP1 leads to extensive plasma membrane ruffling, macropinosome formation and increased uptake of unmodified LDL compared to untreated controls. Subsequently, they demonstrated that TSP1 binds to a specific protein receptor, CD47, to carry out this process.
While these findings were new and exciting, the team sought to uncover the remaining parts of the chain involved in transmitting the instructions to initiate macropinocytosis. Dr Csányi and colleagues genetically blocked the activity of different molecules in the pathway to see whether they are involved in TSP1-CD47-induced membrane ruffling and macropinocytosis. It has previously been shown that TSP1 signals through Nox1, another isoform of the NADPH oxidase family, to induce superoxide anion production in the vascular smooth muscle. Based on these findings, Dr Csányi proposed that TSP1 may use the same mechanism in macrophages to stimulate macropinocytosis. Indeed, they measured increased levels of superoxide anions inside macrophages upon TSP1 treatment. Additionally, macrophages exhibited reduced membrane ruffling, macropinocytosis activity and lipid uptake in response to TSP1 when Nox1 was inhibited. The in vivo importance of this pathway was recapitulated in a mouse model of atherosclerosis.
However, the final piece of this signalling puzzle was still missing. How does the TSP1/CD47/Nox1 axis trigger membrane ruffling and macropinocytosis? Similar to the previous study, the link was cofilin. TSP1 was found to induce cofilin activation when CD47 and Nox1 were functioning in wild-type macrophages but failed to do so when CD47 and Nox1 had been genetically blocked. The study by Dr Csányi and his team demonstrated for the first time that a signalling pathway involving CD47, Nox1 and cofilin contributes to TSP1-induced macropinocytic uptake of unmodified LDL by macrophages.
What Does It All Mean?
Dr Csányi’s work has given us important new insights into the specific mechanisms involved in macrophage uptake of LDL. In addition, it has clarified that macrophages also use macropinocytosis to internalise unmodified forms of bad cholesterol, contributing to the formation of foam cells that become part of atherosclerotic lesions. Dr Csányi states that, ‘these findings support a new paradigm in which redox signaling-mediated lipid macropinocytosis independent of extracellular lipid oxidation propagates initiation and development of vascular inflammatory disease.’ Finally, and perhaps most importantly, this research has highlighted specific mechanistic components that could be targeted for atherosclerosis prevention and treatment.
Meet the researcher

Dr Gábor Csányi
Vascular Biology Center Department of Pharmacology & Toxicology
Augusta University
Georgia, USA
Dr Gábor Csányi graduated from the University of Szeged in Hungary with a Masters of Pharmacology in 2003. He completed his PhD in Medicine at the University of Szeged and Jagiellonian University in Kraków, Poland in 2008. During his PhD, much of his work was focused on characterising functional alterations of endothelium-derived vasodilators that occur in several cardiovascular pathologies, including atherosclerosis, ischemic heart failure, dilated cardiomyopathy and diabetes mellitus. Following his PhD, Dr Csányi moved to the USA to carry out his postdoctoral training in the laboratory of Dr Patrick Pagano at the University of Pittsburgh in Pennsylvania. In his second year, Dr Csányi received an American Heart Association (AHA) Postdoctoral Fellowship. At present, he is an Assistant Professor in the Vascular Biology Center at Augusta University in Georgia, USA and his research is funded by the NIH Pathway to Independence Award (K99/ R00).
CONTACT
E: gcsanyi@augusta.edu
T: (+1) 706 721 1437
W: http://www.augusta.edu/centers/vbc/csanyi.php
KEY COLLABORATORS
Patrick Pagano, PhD, Vascular Medicine Institute, University of Pittsburgh, USA
Stefan Chlopicki, PhD, Jagiellonian Centre for Experimental Therapeutics, Krakow, Poland
David Fulton, PhD, Vascular Biology Center, Augusta University, USA
FUNDING
National Institutes of Health (K99HL114648, 4R00HL114648-03, R01HL079207 and R01HL112914).
LABORATORY
Gabor Csanyi, PhD
Pushpankur Ghoshal, PhD
Bhupesh Singla, PhD Huiping
Lin Madison Carpenter
REFERENCES
G Csányi, DM Feck, P Ghoshal, B Singla, H Lin, S Nagarajan, DN Meijles, IA Ghouleh, N Cantu-Medellin, EE Kelley, L Mateuszuk, JS Isenberg, S Watkins and PJ Pagano, CD47 and Nox1 mediate dynamic fluid-phase macropinocytosis of native LDL, Antioxidants & Redox Signaling, 2017, 1–16.
P Ghoshal, B Singla, H Lin, DM Feck, N Cantu-Medellin, EE Kelley, S Haigh, D Fulton and G Csányi, Nox2-mediated PI3K and cofilin activation convers alternate redox control of macrophage pinocytosis, Antioxidants & Redox Signaling, 2016, 1–15.
![]()