
Dr Jing He – Using Advanced Computational Techniques To Derive Protein Structures From 3d Cryo-Electron Microscopic Images With Insufficient Resolution
Scientist Dr Jing He and her colleagues at Old Dominion University in Virginia use advanced computational techniques to interpret 3-dimensional electron microscopic images of frozen proteins to determine their 3-dimensional structures.

The World of Molecular Structures
Proteins contribute to all biological functions in cells. An average protein is about a few nanometers in size. What do they look like and how do they work? Our understanding of molecular structures has been improved tremendously over the last half a century. The first molecular structure was proposed by American biologist James Watson and the English biophysicist Francis Crick for the DNA molecule in 1953. The first highresolution protein structure was determined for myoglobin by John Kendrew and Max Perutz in 1958. Now, there are over 120,000 molecular structures determined and archived in the Protein Data Bank (PDB) that is publicly accessible worldwide. These molecules were extracted from many different organisms and each has an important role in a biological process. In order to understand the mechanism of a biological process, 3-dimensional structures of molecules are absolutely necessary, because the 3-dimensional structure provides a snapshot of all the details about the molecule. A molecular structure lets you see the details about the molecule. You get to see the locations of all major atoms and how they are related. With modern sciences, it is often possible to figure out what molecules are involved in a biological function. However, it is very hard to figure out how it happens. Scientists want to figure out how molecules interact with each other so that they can design tricks to alter or to control the interaction in curing various diseases.
‘My work is to develop advanced computational methods and tools to interpret 3-dimensional cryo-EM images with insufficient resolutions’
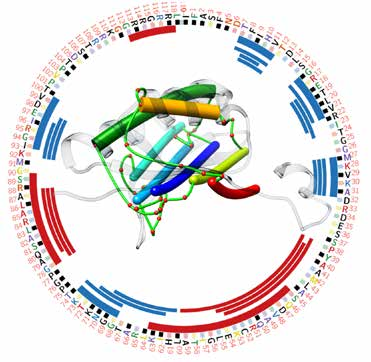
Matching secondary structures (alpha helices (thicker sticks) and beta-strands (thinner sticks)) detected from cryo-EM density map (extracted from EMDB-1780) and those (red and blue bars) predicted from protein sequence.
The Challenge Of Large Molecular Complexes And Cryo-Electron Microscopy
Rapid determination of molecular structures lies in the development of two important techniques: X-ray crystallography and Nuclear Magnetic Resonance. In order to take pictures of small molecules you cannot use visible light – you need to use radiation of small wavelengths, such as X-rays. For X-ray crystallography to be useful, proteins need to be lined up in an ordered array, or a crystal, which can occur under right conditions in the solution. Once good quality crystals are formed, the positions of atoms can be calculated from the X-ray diffraction patterns. Although these two techniques are mainly used in the structure determination of molecules, large molecular complexes and membrane-bound proteins are particularly challenging for these techniques. Large complexes such as ribosomes and various viruses may have tens to thousands of molecules bound together. In general, it is hard to grow crystals for large complexes and membrane-bound complexes. Structural knowledge of large molecular complexes is important, since the structure of large complexes reveals detailed relationship among multiple molecules. Each biological function is performed by multiple molecules. The inter-relationship between them provides important information regarding how the biological function is performed. Cryo-electron microscopy (cryo-EM) is a powerful technique for determining the structures of large molecular complexes. This technique is not limited by the crystallisation of proteins, for which the size of the protein complex matters a lot. The idea behind this technique is to produce many 2-dimensional images of the molecular complex using an electron microscope. As our experience of taking a picture using a camera tells us, the object needs to be still to get a good picture. Thus, in this technique, molecules are fixed in ice by plunging them in a liquid nitrogen bath. But how are two-dimensional pictures converted into a 3-dimensional image? Suppose that we want to know the 3-dimensional volumetric image of a car in great details using a camera that can only produce 2-dimensional images of the car. We can randomly lay many copies of the same car in a parking lot and take pictures of all cars. Each 2-dimensional image of the car contains information about the car from a particular angle. Given sufficient such images, it is possible to computationally merge them into a 3-dimensional volume of the car that agrees, in principle, with all 2-dimensional pictures. Cryo-EM techniques have improved dramatically over the last twenty years and it is only recently that the determination of atomic-resolution structures is possible for many molecular complexes. As of August 2016, there have been 1134 atomic structures of molecular complexes resolved using cryo-EM techniques and they are all deposited in PDB.

3.8Å resolution image (extracted from EMDB-5199)
Figuring Out the Structure from 3D Images with Insufficient Resolutions
In order to derive atomic structures using cryo-EM, a 3-dimensional volumetric image, or so-called density map, must be interpreted. If the density map is produced at a high-resolution of around 3Å, then their atomic structures can be resolved, since sufficient details about atoms can be figured out from such images. When most components of a car are distinguishable from an image, it is easier to tell the entire structure of the car. Similarly, the location of most major atoms can be figured out directly from such a high-resolution density map.
The lower the resolution of the image, the harder it is to figure out the structure of the protein. If your specimen only allows you to obtain data at medium resolution, about 5–10Å say, then less information is obtained from such images than from the high-resolution images. This is similar to the problem of drawing the original car based on the car in the junk yard. Molecules are delicate, and it is challenging to obtain high-resolution images for many molecules. A method to derive atomic structures from medium resolution images will allow us to push the limits of structure determination for those molecules that are more dynamic and only have density maps with insufficient resolution. Dr He and her colleagues’ specialty is using advanced computational techniques to combine both the volumetric data and protein sequence data to figure out the structure of the protein from a ‘cryo-EM density map with 5-10Å resolution.
 Attacking Two Critical Steps in Interpreting Cryo-EM 3D Images
Attacking Two Critical Steps in Interpreting Cryo-EM 3D Images
To make sense of a sub-standard image of a car, two critical steps are needed: to distinguish the individual components as much as possible, and to make connections between the components in order to set up the framework of the car. The individual components seen may have errors, and therefore a framework needs to be created with the consideration of such uncertainty. Dr He looks at this problem as three different objectives directed at the same general goal, in describing the structure of large molecules. First, she wants to improve the accuracy in secondary structure detection from cryo-EM density maps at medium resolutions. ‘The most characteristic components in the mediumresolution density maps are secondary structures such as ạ-helices and ß-strands,’ she explains. The accuracy of secondary structure detection plays an important role in the determination of the tertiary structure of the protein. The ạ-helix is a compact helical structure formed by a polypeptide chain. They have the right sequence of amino acids to allow a close, stable curling of the coils. A ß-strand is a segment of chain that is ‘stretched’, and when multiple ß-strands come together, they form a sheet-like shape called a ß-sheet. You might imagine a helix as a piece of traditional telephone wire, and a ß-sheet as multiple ladders laid out and tied together with soft strings, where each half of a ladder is a ß-strand. It is amazing how the molecular world is not actually that chaotic at all. The same insight used to stabilise objects in the real world is also used to stabilise protein structures.
Dr He also wants to develop computational methods that derive the topologies – a mathematical term used to describe the framework of the car – of secondary structures when inaccurate data is used in the first place. Finally, she wants to turn the complex computational methods that she develops into user friendly tools that anyone can use, even someone that is not a trained mathematician or computer scientist.
Computation Makes You See that You Do Not See
Addressing her first objective, Dr He and her co-workers and students have developed computational methods to detect ạ-helices and ß-sheets using their characteristic shape properties. Computer programs are written to search the 3D image of a protein for cylindrical shape regions as helices and plane-like regions asß-sheets. This process is called pattern recognition and is a powerful technique to distinguish objects as long as they have good characteristics and the image is of sufficient quality. Generally, if your eyes can distinguish an object from the background, computer programs can be designed to find it automatically. However, what happens when your eyes cannot see the object? Would it still be possible to detect it computationally? This question puzzled Dr He for a long time until she and her student found a way to push pattern recognition beyond its capability by introducing modelling into the problem. Since the spacing between two neighbouring ß-strands is about 4.5–5Å, they can’t be resolved in a 3D image of 5–10Å resolution. When we can’t see ß-strands, is it still possible to detect them? Dr He and her student Dong Si discovered that the arrangement of ß-strands is linked to the twist of the ß-sheet. It had been discovered in the 70s that all ß-sheets exhibit righthanded twisting, which means that they are not flat. They utilised the asymmetric nature of the ß-sheets to model ß-strands. This work demonstrated that it is possible to extract the location of ß-strands from medium resolution cryo-EM images. The location of ß-strands is one of the two treasures existing in medium-resolution images. This further justifies the value of images at medium resolutions. ‘What we learned is that advanced computational methods are powerful in detecting objects that are not resolved in certain cases.’ Dr He explains. This work is included in StrandTwister, a method to predict the trace of ß-strand from ß-sheet image at medium resolutions.
Machine Learning to Utilise Large Amounts of Data
As more 3D cryo-EM images and their corresponding atomic structures are deposited in the public database Electron Microscopy Data Bank (EMDB) and PDB, it is possible to involve machine learning techniques in pattern recognition of secondary structures. Machine learning techniques uses existing data to train a computer program so that it remembers the patterns and will be able to recognise similar patterns automatically when they appear in a new set of data. Dr Jing He, Dr Dong Si (previously a student of Dr He) and their collaborator Dr Shuiwang Ji developed a support-vector-machine (SVM)-based learning method to detect both ạ-helices and ß-sheets from cryo-EM maps. Recently they developed a deep learning approach using a convolutional neural network (CNN) for the same problem. Advanced computational techniques such as modelling and machine learning have enhanced the capability of pattern recognition because they allow us to explore patterns beyond the current image. Those weak patterns in the current image may become more evident when other alternative patterns are modelled or when other images are brought into the analysis.
All of this work dramatically improves Dr He’s ability to determine secondary protein structure in cases of medium resolution cryo-EM data. But that was only the first of her three objectives.
Good Algorithms Mean Less Effort Dealing with Errors in Data
Deriving structures from 3D images with insufficient resolutions requires effort in modelling uncertainty or possible errors in the data. Using the analogy of figuring out the framework of the car, we need to figure out how the protein chain threads through the 3D image. You can imagine a protein as a chain of beads (or a necklace), where each bead resembles an amino acid. A helix/ß-strand is in fact a segment of the chain often consisting of 3 to 30 consecutive amino acids. The secondary structures on the protein sequence show their identity but not their location in 3-dimensional space. The secondary structures detected from the cryo-EM 3D image provide their location but not identity. The idea is to combine the 3D image and the protein sequence to get both the identity and spatial location of secondary structures. Since secondary structures are major components of a structure, once they are figured out, the entire framework of the structure can be derived. A naïve way of combining the protein sequence with the 3D image involves trying out all possible ways to thread the sequence through the image. This means huge computation since the computational time becomes exponential as the number of the alternatives and the number of components increase. Dr He’s team, including her collaborators Dr Desh Ranjan, Dr M. Zubair, and Dr Abhishek Biswas (previously a student of Dr Ranjan and Dr Zubair), developed a smart way to try out alternatives with significantly reduced computation using what is called a dynamic programming algorithm. To give an example how much a good algorithm can cut down computational effort, using a naïve way to try out all possible alternatives takes 493718.75 seconds, but only 22.35 seconds using the dynamic programming method developed for the same task. A good method needs to be implemented in a userfriendly tool before it is actually useful. Dr He and co-workers are trying to provide researchers with secondary structure detection and analysis tools that are easy to use.
Riding the Wave into the Future
Dr He explains that she has always been curious about biology: ‘It is a field with so many unanswered questions, and computation has become such an important component of biology.’ And now, modern biology has become quite dependent on Dr He’s specialty of mathematical computation and computer science. ‘Cryo-EM has evolved dramatically over the last twenty years. Very few people used to believe in it, yet now many people want to use it, since it is becoming a mature and powerful technique for structural determination of large biological complexes.’ And now, Dr He is riding the wave of progress into the future. When asked what the future holds for her research, Dr He says: ‘advanced computational methods have been shown to cut down the computational time and to enhance the accuracy in interpreting medium-resolution cryo-EM maps. I believe that integrating advanced computational methods deeply in biological problems is the future approach for complex biological problems.’
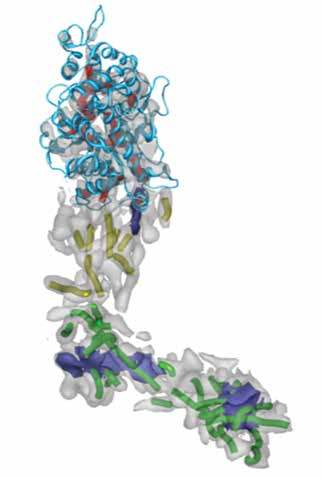
Herpes Virus VP5 protein
Meet the researcher

Dr Jing He
Associate Professor
Department of Computer Science
Old Dominion University
Virginia, USA
Dr Jing He received her BSc in Applied Mathematics in 1990 from Jilin University in China. She received an MSc in Applied Mathematics in 1994 from New Mexico State University, and was then awarded her PhD in Structural & Computational Biology & Molecular Biophysics from Baylor College of Medicine, in Houston, Texas, in 2001. She joined Department of Computer Science at New Mexico State University in 2002 and then Old Dominion University in 2009.
Dr He’s research interests include developing computational approaches to derive the structure of proteins from data obtained by cryo-electron microscopy. At a medium resolution such as 5–10Å, it is extremely difficult to determine protein structure directly from the volumetric data, so she tries to develop computational methods and tools in order to automate the determination of protein structures from the cryo-electron microscopy density maps. Dr He has authored and co-authored more than 50 papers in peer-reviewed journals and scientific conferences dealing with cryo-electron microscopy imaging and bioinformatics, as well as several book chapters.
CONTACT
T: (+1) 757 683 7716
E: jhe@odu.edu
W: http://www.cs.odu.edu/~jhe/
FUNDING
National Science Foundation, Advances in Biological Informatics
CO-PRINCIPLE INVESTIGATORS
Dr Mohammed Zubair, Computer Science Department, ODU
Dr Desh Ranjan, Computer Science Department, ODU
Shuiwang Ji, Computer Science Department, ODU until 2015, currently at Washington State University at Pullman
REFERENCES
D Si and J He, Tracing Beta Strands Using StrandTwister from Cryo-EM Density Maps at Medium Resolutions, Structure, 2014, 22, 1–12.
D Si, S Ji, K Al Nasr, J He, A machine learning approach for the identification of protein secondary structure elements from electron cryo-microscopy density maps, Biopolymers, 2012, 97, 698–708.
A Biswas, D Ranjan, M Zubair, S Zeil, KA Nasr and J He, An Effective Computational Method Incorporating Multiple Secondary Structure Predictions in Topology Determination for Cryo-EM Images, IEEE/ACM Trans Comput Biol Bioinform., 2016. DOI: 10.1109/TCBB.2016.2543721
A Biswas, D Ranjan, M Zubair and J He, A Dynamic Programming Algorithm for Finding the Optimal Placement of a Secondary Structure Topology in Cryo-EM Data, Journal of Computational Biology, 2015, 22, 837–843.

Download

{kind=link}