
Dr Zhihua Jiang – RNA and the Key to the Diversity of Life
Over 90% of a mammalian genome is transcribed into RNA molecules, including both protein-coding and non-coding transcripts. Genes often produce one or more expressed isoforms. Dr Zhihua Jiang at Washington State University has developed novel next-generation sequencing techniques that can be used to understand how RNA diversity occurs at the cell, tissue and whole-body levels and how this diversity contributes to the development and growth of animals into different forms or phenotypes.
The Origin of Phenotypes
The central dogma of molecular biology states that, ‘DNA (the genome) makes RNA (the transcriptome) that makes protein (the proteome).’ However, research has clearly indicated that many biological systems are far more complex. From the beginning of life with one fertilised egg to the end with 1012–1016 cells in the human body, the genome remains relatively static. However, the shape and form of the body, or the ‘phenome’ (the collection of all an organism’s phenotypes), changes dynamically in response to diverse functions.
How can a finite genome produce an infinite phenome during a life-span? The key to answering this question is understanding how alternative RNA transcripts are produced from genes. We know that alternative transcripts coordinate information flow from genome to phenome in response to internal, external, universal and biotechnological environments. Indeed, a genome harbours a large number of DNA variants, most of which are silent. The only DNA mutations that may affect phenotypes include those located in regions that change protein expression or truncate proteins. In brief, functional DNA variants are those that potentially affect RNA transcription or structures. Recent research clearly points to the key role of RNA variants in the regulation of genome functions, but their contributions to the origin of phenotypes remain largely undetermined.
Generally speaking, RNA variants result from various mechanisms. Timely use of transcript variants is essential for cell maturation (differentiation) and development, and response to environmental challenges. The misuse of alternative transcripts often causes defects, diseases and disorders that affect nearly every body system. As such, alternative transcripts can be used as biomarkers to improve early diagnosis of diseases, conditions and even cancers. They can also be used as targets for novel treatment options.
The Analysis of RNA Variants
There is a number of different types of RNA that are classified as coding and non-coding due to their diverse functions in molecular biology. Transitory mRNAs (messenger RNAs) aided by tRNA (transfer RNAs) and ribosomes containing ribosomal proteins and rRNA (ribosomal RNAs) are responsible for the synthesis of proteins. Small nuclear RNAs (snRNAs) and snoRNAs (small nucleolar RNAs) are often involved in either mRNA or rRNA processing, while miRNAs (microRNAs) degrade mRNAs and even silence gene function at the DNA level. Long non-coding RNAs (lncRNAs) are another type of RNA whose functions remain largely uncharacterised.
E-book

Reference
https://doi.org/10.26320/SCIENTIA312
Share
{kind=link}
‘The WTTS-seq assay serves as a powerful tool for the research community to investigate transcriptomes and reveal poly(A) site usages specific to complex phenotypes, disease stages, or biological processes in humans, animals, and plants.’
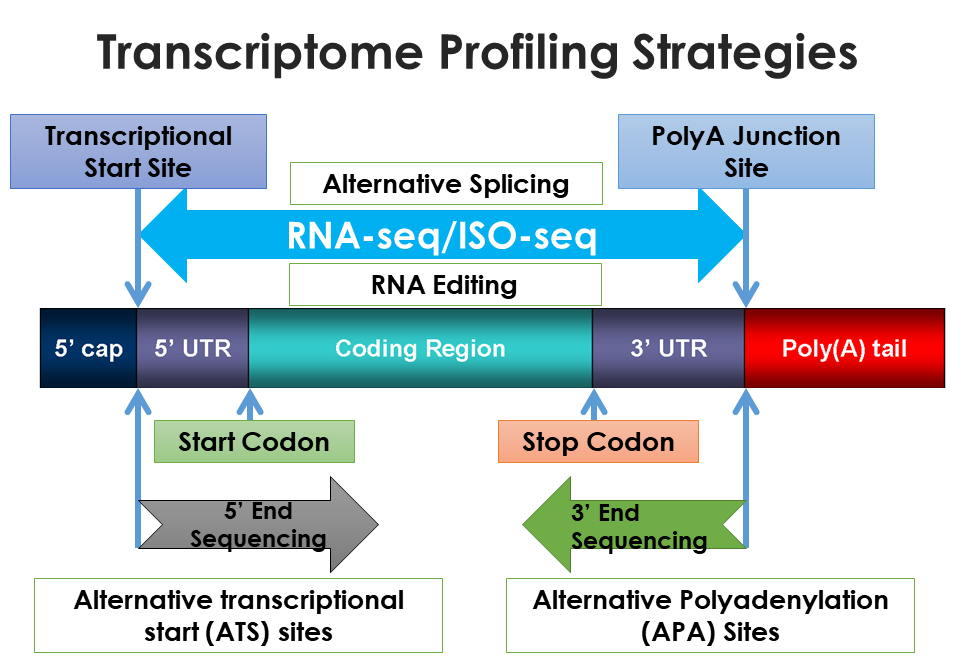
Regardless of type, RNAs are usually converted in the laboratory into complementary DNA (cDNA) for sequencing. Nucleic acid sequencing determines the precise order of nucleic acids within a DNA or RNA molecule and establishes how amino acids are formed into proteins. DNA was first sequenced in the early 1970s and advances in technology have allowed scientists to decipher the entire genetic code of many different organisms faster and more efficiently. In the late 1990s, there were major advances in the speed at which machines could carry out sequencing reactions, as well as the length of the fragments that could be sequenced. In order to sequence RNAs from the transcriptional start site to transcriptional stop site, both next and third generation sequencing platforms have been developed. Basically, these methods were much quicker than previous sequencing methods.
RNA sequencing (RNA-seq) is a next-generation sequencing method that provides short reads, while isoform sequencing (Iso-seq) as a third-generation sequencing method that produces long reads for transcriptome analysis. Because research studies revealed that transcriptome diversity is largely due to the use of alternative transcription start and termination sites, Dr Zhihua Jiang and his group at Washington State University have focused on the development of methods to capture both ends of RNA transcripts.

A New Way to Sequence
Dr Jiang and his team of scientists have developed two cutting-edge techniques, known as whole transcriptome start site sequencing (WTSS-seq) and whole transcriptome termini site sequencing (WTTS-seq). These techniques offer a number of improvements over existing RNA sequencing technologies such as RNA-seq, producing sequences that cover the entire transcriptome in cells, tissues, organs and the whole body.
The techniques developed in Dr Jiang’s lab have a number of advantages over current RNA-seq techniques. For example, WTTS-seq can easily detect the alternative polyadenylation (APA) sites that mark the end of a transcript, which is difficult to determine with RNA-seq. The WTTS-seq method is also at least 67% cheaper than RNA-seq. Other challenges with RNA-seq include the under-representation of shorter fragments or RNA molecules in the final sample. Short fragments are easily lost, resulting in larger fragments dominating the sample. Bias can also occur when one or a few transcripts in a given sample are expressed at extremely high levels, thereby downplaying the number of times other mRNA molecules are identified or read.
RNA-seq often requires 10–20 times more reads than a typical sequencing method, thus creating challenges in analysis and computational methods due to the large amount of data that needs to be stored, retrieved and processed. In addition, RNA-seq cannot sufficiently detect genes or transcripts with low levels of expression. The process also remains inefficient due to the many steps involved in preparing the sequencing libraries. Dr Jiang stresses, however, that both WTTS-seq and RNA-seq can be used together to avoid further validation using other methods.
Requiring only a minute amount of RNA, the WTTS-seq method starts with fragmentation or separation of total RNA, followed by enrichment of regions of polyadenylation, called poly(A)tails, located at the end of RNA fragments and by PCR (polymerase chain reaction) to amplify products for size selection. After these steps, the fragments can be analysed by the Ion Torrent, a popular sequencing platform.
Recent examination of an online database revealed thousands of APA sites harboured in the genomes of humans, mice, rats and chickens that mark the end of transcripts. Using the whole transcriptome termini site sequencing (WTTS-seq) method, Dr Jiang first used a frog genome to demonstrate how the WTTS-seq method can unravel the APA patterns across diverse developmental stages.
The Rules of Life via Alternative Polyadenylation
Using tissue collected from the frog species Xenopus tropicalis, Dr Jiang compared RNA transcripts between sexes in addition to their ages. The APAs were incorporated at different sites for males and females at similar ages. The researchers also observed striking differences in the use of APA sites on maternal and offspring RNA and between embryos and adults. Unravelling these differences in APA patterns generates a wealth of knowledge that can be used to predict how animals within the same species might express genes differently.
After the material was sequenced, statistical, visual and functional tools were used to study APA events. The team found striking differences in the use of APA sites between genes that went on to make protein and those that did not. Most coding genes had more than one APA site, with protein-coding genes containing an average of 5.87 APA sites per gene. Usage of APA sites was different between adult males and females, with young males using thousands fewer APA sites than young females. The researchers also examined the APA sites of older adult animals using their WTSS-seq technique. At younger ages, females had more APA sites that coded for protein than males, but the trend was reversed in the older age group.
In another study, the group used WTTS-seq technology to study APA events in mice with a gene that had been ‘knocked out’ or prevented from making protein, compared to mice that made the protein normally. These results indicated that both APA- and gene-based analyses produce very different results. In particular, the gene-based approach could not explain the changes in muscle development between the two animal models. The gene-based analysis had much less power than the APA-based analysis.
These results identified APA sites that were expressed differently in mice with the original gene intact. These differently-expressed APA (DE-APA) sites were located on the mRNA (that makes protein) in areas near the centre of the molecule and generated above-average protein levels. The researchers found that the DE-APA sites that generated lower levels of protein had APA sites located mostly in conventional locations, closer to the edges of the RNA molecule. They concluded that gene knockout may affect APA events, resulting in differences in the tissues of the mouse as it grows and develops, in this case, muscle.
Dr Jiang also recently published research in which his research group found distinct APA patterns associated with repair and inflammation of the uterus in healthy and unhealthy cows using WTTS-seq. Here, WTTS-seq also revealed essential microRNA genes that are involved in inflammation and anti-inflammation processes and dysregulated APA events in unhealthy cows in comparison to healthy cows.

Moving Forward
Sequencing technology is rapidly advancing, and Dr Jiang believes the timing is right for the research community to assemble the whole transcriptomes of all species. ‘This analysis is also important because it provides the first steps toward functional characterisation and annotation of genes/genomes previously revealed by DNA sequencing,’ says Dr Jiang. ‘The WTTS-seq assay serves as a powerful tool for the research community to investigate transcriptomes and reveal poly(A) site usages specific to complex phenotypes, disease stages, or biological processes in humans, animals, and plants.’
In addition to WTTS-seq, his novel WTSS-seq method is also a sensitive and powerful technology that can be used to show how information flows from DNA (genome) to RNA (transcriptome) to protein (proteome) and can be used to explain the variety of altered physical outcomes (or phenotypes) seen in animal development in a number of different environments. The group has filed a provisional patent for their WTSS-seq method and Dr Jiang hopes that the technology can trigger interest from the wider scientific community to use these techniques to pursue their own studies. With both WTSS-seq and WTTS-seq, the group also plans to establish their own online resources for recording both alternative transcriptional start sites and termination sites in cattle, chicken, mouse, rat and the Xenopus tropicalis species of frog in the near future.
Meet the researcher

Dr Zhihua Jiang
Department of Animal Sciences
Washington State University
Pullman, WA
USA
Dr Zhihua Jiang is a professor in the Department of Animal Sciences at Washington State University and a Hatch Program Chair in Animal Biology and Biomedicine. He completed his PhD at the University of Zagreb in Croatia and was a postdoctoral fellow at the Roslin Institute, University of Edinburgh, Scotland and the National Center for Scientific Research (CNRS), University of Rennes 1, France. Previously he was a research professor at the University of Guelph in Canada in the Department of Animal and Poultry Science and a professor in the Department of Animal Science at Nanjing Agriculture University in China. His current work focuses on studying the origin of phenotypes using state of the art genomics and transcriptomics approaches in combination with bioinformatics and cell/embryo biology.
CONTACT
E: jiangz@wsu.edu
W: https://ansci.wsu.edu/people/faculty/zhihua-jiang/
KEY COLLABORATORS
Dr Richard M. Harland at UC Berkeley, Dr Pablo J. Ross at UC Davis and Dr Jon F. Davis at Washington State University
FUNDING
National Institute of Health
United States Department of Agriculture
The State of Washington Initiatives
FURTHER READING
Z Jiang, X Zhou, R Li, JJ Michal, S Zhang, Z Zhang and RM Harland, Whole transcriptome analysis with sequencing: methods, challenges and potential solutions, Cellular and Molecular Life Sciences, 2015, 72, 3425–3439.
X Zhou, R Li, JJ Michal, XL Wu, Z Liu, H Zhao, Y Xia, W Du, MR Wildung, DJ Pouchnik, RM Harland and Z Jiang, Accurate profiling of gene expression and alternative polyadenylation with whole transcriptome termini site sequencing (WTTS-Seq), Genetics, 2016, 203, 683–697.
JN Brutman, X Zhou, Y Zhang, J Michal, B Stark, Z Jiang and JF Davis, Mapping diet-induced alternative polyadenylation of hypothalamic transcripts in the obese rat, Physiology and Behavior, 2018, 188, 173–180.
SW Zhang, YZ Zhang, X Zhou, X Fu, JJ Michal, GL Ji, M Du, JF Davis and Z Jiang, Alternative polyadenylation drives genome-to-phenome information detours in the AMPKα1 and AMPKα2 knockout mice, Scientific Reports, 2018, 8, 6462.
YQ Zhang, SA Carrion, YZ Zhang, XH Zhang, AL Zinski, JJ Michal and Z Jiang, Alternative polyadenylation analysis in animals and plants: newly developed strategies for profiling, processing and validation, International Journal of Biological Sciences, 2018, 14, 1709–1714.
X Zhou, Y Zhang, JJ Michal, LJ Qu, SW Zhang, MR Wildung, W Du, DJ Pouchnik, H Zhao, Y Xia, HH Shi, GL Ji, JF Davis, GD Smith, MD Griswold, RM Harland and Z Jiang, Alternative polyadenylation coordinates embryonic development, sexual dimorphisms and longitudinal growth in Xenopus Tropicalis, Cellular and Molecular Life Sciences, 2018 (In press).
MJ Stotts, YZ Zhang, SW Zhang, JJ Michal, JS Velez, B Hans, M Maquivar and Z Jiang, Alternative polyadenylation events in epithelial cells sense endometritis progression in dairy cows (In preparation).
![]()