
Professor Elizabeth Jonas – The Mitochondrion: The Powerhouse Behind Neurotransmission
Professor Elizabeth Jonas and her colleagues at Yale University study the function of cell components called mitochondria and their role in neurotransmission. In particular, Professor Jonas is interested in characterising how channels in the mitochondrial membrane affect neuronal function during processes like memory formation and learning, and how they enhance or reduce neuronal viability during disease.

Neurotransmission – firing on all cylinders.
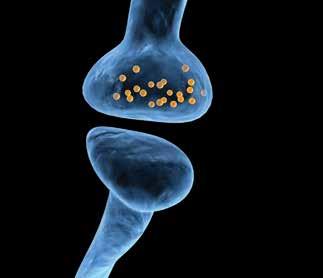
Neurons form an incredibly complex network throughout our bodies, and play a fundamental physiological role. Neurons form the link between brain and body by transmitting information to and from the brain and spinal cord to peripheral tissues such as our muscles. The brain can encode information as patterns of neural impulses that pass from neuron to neuron. Neurons also comprise the brain and spinal tissue itself and normal neuronal function forms the basis for a diverse set of processes including thought, memory formation and movement. Neurotransmission is the procedure by which neuronal cells communicate with each other. Typically, neurons do not directly touch each other, but rather exchange information at specialised structures surrounding a very thin gap between neurons called a synapse. At a synapse, an activated neuron, through which a neural impulse has travelled, will release molecules called neurotransmitters which can travel across the synaptic cleft and bind to receptors in a second neuron. In this manner, the neural impulse can propagate along the second neuron and continue on its way. As you might have guessed, this process requires energy, and alterations in the mechanism by which this energy is provided can have profound effects on neuronal function and viability, and implications in disease.
The nuts and bolts of neurotransmission: the mitochondrial system.
Much of the energy demands of neurotransmission are met by a small organelle present in neurons called the mitochondrion. Mitochondria are membranebound structures which produce a substance called adenosine triphosphate (ATP), which is used by the neuron as a source of energy during neurotransmission. Mitochondrial efficiency refers to how much ATP is produced per molecule of glucose or oxygen taken up by a cell or organism, and when this process is inefficient less ATP is produced. Neurotransmission also involves the uptake of calcium through small channels or pores in the neuronal membrane. Calcium facilitates the release of neurotransmitters from the neuron. However, once neurotransmission is complete, the calcium that entered the neuron needs to be cleared, to allow the system to reset. Mitochondria play a role in helping to mop up this calcium through an intricate system of channels in their membranes. They also re-release the sequestered calcium and so not only do they help to reset calcium levels in the neuron, but they participate in carefully regulating calcium levels during neurotransmission. This process also has important effects on ATP production by the mitochondria and can affect how excitable the neuron is. If the calcium-regulating or ATP-producing processes become altered this can change the excitability of a neuron over time, or can even cause the death of the neuron. In fact, channels in mitochondria are important factors in cell death and may contribute substantially to the permanent alteration in neuronal function in the brain that underlies learning and memory formation and other forms of neural plasticity. These processes may go very wrong in neurodegenerative diseases like Alzheimer’s or Parkinson’s disease; therefore, mitochondrial dysfunction is heavily implicated in these diseases.
‘We think we have found a key molecule that forms a major cell death-inducing mitochondrial ion channel’
How is this process regulated?
Professor Elizabeth Jonas and her team are interested in the regulation of ion transport across the mitochondrial membrane, with a view to understanding the role of the mitochondrion in normal activities such as learning and memory, as well as its role in disease processes which result in abnormal neuronal activity or neuronal cell death. The team focuses on identifying the most important molecular players in the energy and calcium dynamics of neurotransmission and on studying how these interact, like filling in pieces of a jigsaw puzzle. In fact, Professor Jonas is credited with developing a completely new technique to study ion channels inside living cells, using two electrodes, whereby one electrode is sheathed inside another and is used to probe for ion channels in the membranes of organelles, like mitochondria. Interestingly, her initial goal when applying this technique was to find ion channels on the membranes of large secretory granules in invertebrate neurons. She was surprised when instead, she discovered calcium release channels in mitochondria, a discovery which sparked her intense interest in mitochondrial channels and the role they play in disease. Understanding what substances pass in and out of the mitochondrion, when they move in and out, and what molecules permit or block their movement and why, are key elements of Professor Jonas’ research. For example, the team discovered that a segment of an enzyme called ATP synthase, which is involved in the production of ATP in the mitochondrion, functions as a channel in the inner mitochondrial membrane, where it is embedded. Professor Jonas believes that in this discovery, the team has uncovered the identity of a very important channel, called the mitochondrial permeability transition pore. The channel can open and close, allowing mitochondrial contents to travel through it. Exposure to high concentrations of calcium results in permeability transition pore opening which may act physiologically in the neuron to help re-release sequestered calcium, but, if open for a longer time, may also disrupt mitochondrial function. These events ultimately lead to loss of normal neurotransmission and neuronal death. In addition, the team is interested in other molecules that can interact with, and affect the behaviour of mitochondrial pores or otherwise modulate mitochondrial activity and neurotransmission. The Bcl-2 family of proteins, originally identified in cancer cells, is involved in the regulation of mitochondrial membrane permeability among other processes. Several members of the family tend to promote cell death, while others prevent cell death. This comprises a particularly important function for cancer cells, which rely on anti-death Bcl-2 family members to promote their cancer-inducing behaviour. In neurons the anti-death Bcl-2 family members prevent death due to many forms of injury or aging, and this function in neurons is termed neuroprotection. One member of the family is called Bcl-xL. Bcl-xL spans the mitochondrial membrane and its classical role is to prevent other members of the Bcl-2 family from initiating a cell death program known as apoptosis. However, Professor Jonas’ team has found that Bcl-xL can also improve the efficiency of mitochondrial function during neurotransmission by interacting with the ATP synthase enzyme and the mitochondrial permeability transition pore. Jonas’ team finds that Bcl-xL essentially contributes to prevention of leaking of H+ ions through the mitochondrial permeability transition pore, which improves the ability of the mitochondria to increase ATP production, while reducing the metabolic demands on mitochondria. These processes may partially underlie the potential of Bcl-xL to protect neurons from degeneration and may also play a role in memory formation in the brain.
Synaptic plasticity
So how do these complex phenomena on a small scale in neurons relate to appreciable effects in human beings? One mechanism these factors can affect is synaptic plasticity, which is the tendency of a synapse to become stronger or weaker over time, as a result of changes in its activity. It is hypothesised that this process could underlie our ability to create memories and retain learned information, in addition to potential involvement in pathologies like addiction, depression, autism, Alzheimer’s Disease and in movement disorders such as Parkinson’s Disease. Mitochondrial ion channels play a role in synaptic plasticity, through changes in the uptake or re-release of calcium into the neuron, which can enhance or suppress neurotransmitter release into the synapse, but also perhaps through long lasting changes in the efficiency of energy production over time (mitochondrial plasticity). In these ways, increased levels of Bcl-xL can enhance the potential of a neuron to release neurotransmitter over the long-term. Therefore, changes in the activity of mitochondrial ion channels could potentially play a significant role in brain development and in preventing brain aging.
Cell death
In addition to changing levels of neuronal activity, mitochondrial ion channels also control cell death. In diseases such as stroke, the blood supply to an area of the brain is cut off by the blockage of a blood vessel feeding the brain. The neurons in the affected area are deprived of nutrients and oxygen and die, potentially leading to impairments in cognition and mobility, including paralysis. Some of the affected cells undergo a process called apoptosis, during which a complex biochemical chain of events causes them to be destroyed. This process involves mitochondrial ion channel activity and Bcl-2 proteins. Professor Jonas’ team investigated the role of Bcl-xL in neurons which had been starved of nutrients and oxygen during the process known as neuronal ischemia. They discovered that Bcl-xL was a key component of the apoptotic cascade in this type of cell, and that during stroke or ischemic brain injury, neurons tended to form a slightly different form of Bcl-xL, that induced, rather than protected from, apoptosis. Surprisingly, fighting this pro-death form of Bcl-xL was easy. The group used a drug that was already known to block the anti-apoptotic full length form of Bcl-xL in cancer cells. In the Jonas team hands, the drug, ABT-737, also appeared to effectively interact with the pro-death form of Bcl-xL, significantly reducing the amount of cell death caused by the brain ischemia. The team concluded that Bcl-xL might represent an important drug target in the treatment of diseases like stroke. If a drug could inhibit the pro-apoptotic form of Bcl-xL in stroke patients, then perhaps levels of cell death and functional impairment could be dramatically reduced.
While pro-apoptotic Bcl-xL appears to contribute to neuronal cell death processes in diseases like stroke, full length Bcl-xL may promote neuronal survival under non-ischemic conditions. In fact, Professor Jonas and her team have hypothesised that neuroprotective molecules like Bcl-xL may prevent the onset of neurodegenerative processes and consequently neurodegenerative disease. Conversely, the loss of neuroprotective molecules like Bcl-xL, could permit neurodegenerative processes to occur. Neurodegeneration results in the loss of synaptic function, or in neuronal death, and underlies a host of diseases including Alzheimer’s disease, Parkinson’s disease and Huntington’s disease among others. These conditions are a significant source of mortality and suffering. Consequently, research that could provide answers about the molecular basis for neurodegeneration, such as that undertaken by Professor Jonas, could be very valuable to society.
The next steps for Professor Jonas’ team.
‘We now want to find out exactly how Bcl-xL assists the mitochondria to alter their efficiency during formation of memories in the hippocampus and how Bcl-xL works in other neurons in the brain’ Professor Jonas tells Scientia. The team is also interested in finding out more about how this interaction may go awry in neurodegenerative conditions. These results could aid in finding new drug targets to help in the treatment of such conditions. Future research will look at other proteins that interact with ATP synthase to improve the efficiency of mitochondrial function. One example is DJ1. This protein is highly expressed in the brain and in cancer cells, and is mutated in a familial form of Parkinson’s disease, suggesting a strong link with the pathogenesis of Parkinson’s disease.
In addition to looking at neurodegenerative disease, Professor Jonas also plans to study the role of mitochondrial efficiency in neurodevelopmental disorders. Fragile X syndrome is one such disorder in the category of autism spectrum disease, resulting in intellectual disability. Fragile X disorder is caused by an abnormality in the FMR1 gene which encodes the FMRP protein. ‘Preliminarily, we have found that Bcl-xL and FMRP work together to protect neurons and particularly synapses during brain development’ says Professor Jonas. The FMRP and Bcl-xL combination may work to regulate the efficiency of mitochondrial function and this in turn seems to regulate protein synthesis efficiency during synaptic plasticity. The team suspects that the abnormal FMRP protein may disrupt mitochondrial function and thereby prevent the normal regulation of protein synthesis in Fragile X patients’ brains. These findings could underlie in part the pathogenesis of the disorder.
The implications of the results of this neurological research are applicable to a variety of diverse disease states. The team with Dr Kambiz Alavian at Imperial College, London, have also recently started to determine the role of the ATP synthase channel they have identified as the mitochondrial permeability transition pore, in the growth of cancer. In other studies at Yale, they have begun to characterise the structure of the channel in detail using a technique called cryo-electron microscopy, in order to better understand how it functions.
Meet the researcher

Professor Elizabeth Jonas
Associate Professor
Dept. of Internal Medicine (Endocrinology) and Dept. of Neuroscience
Yale University School of Medicine, New Haven, CT
Professor Elizabeth Jonas is an Associate Professor in the Department of Internal Medicine (Endocrinology) and the Department of Neuroscience at Yale University. After completing a BA at Yale University, Prof. Jonas completed her M.D. at New York University. Subsequently, she completed residencies in Neurology and Internal Medicine at the Yale University School of Medicine and a Postdoctoral Fellowship in Pharmacology at the same location. She began an Assistant Professorship in the Department of Internal Medicine at Yale University before becoming an Associate Professor in the Department of Internal Medicine (Endocrinology) and the Department of Neuroscience at Yale University.
CONTACT
T: (+1) 203 785 3087
W: http://bbs.yale.edu/people/elizabeth_jonas-1.profile
CURRENT LAB MEMBERS
Nelli Mnatsakanyan
Han-A Park
Pawel Licznerski
Paige Miranda
Rongmin Chen
Jing Wu
KEY COLLABORATORS
J Marie Hardwick, Johns Hopkins University, USA
Leonard K Kaczmarek, Yale University, USA
R Suzanne Zukin, Albert Einstein College of Medicine, USA
Kambiz N Alavian, Imperial College, London, UK
Dimitry Ofengeim, Harvard, USA
John Hickman, IMI PREDECT
Valentin Gribkoff, Yale University, USA
Casey Kinnally, NYU, USA
Evgeny Pavlov, NYU, USA
George A Porter, University of Rochester, USA
Keith Nehrke, University of Rochester, USA
Paul Brooks, University of Rochester, USA
FUNDING
NIH/NINDS
FRAXA Research Foundation
Knopp Neurosciences Inc.
GeminX Pharmaceuticals
American Heart Established Investigator
Grass Faculty Award
American Foundation for Aging Research
Hartford Foundation Fellowship
Hellman Fellowship
Frank R. Lillie Fellowship
Ann E. Kammer Memorial Fellowship Fund
National Science Foundation POWRE Program/NSF 97-91
AASH/Glaxo Wellcome Headache Research Award
Diabetes and Endocrine Research Council Award
DOWNLOAD

{kind=link}