
Professor Karima Djabali – Putting A Stop To Premature Ageing
For over ten years, Professor Karima Djabali and her team have been on a mission to find a cure for Hutchinson- Gilford Progeria Syndrome. This cutting edge research can inform both interventions for this fatal disorder and reveal further insights into normal ageing.

Hutchinson-Gilford Progeria Syndrome is caused by mutations on the LMNA gene which encodes lamin A and C proteins that are critical components of the nuclear envelope that protects the genome integrity.
Journey to a cure

Normal Aging
With each study, Professor Djabali and her colleagues get one step closer to a cure. Their 2010 paper explains the influence of protein farnesylation on the pathology of HGPS and how this could be inhibited. Protein farnesylation in this case refers to the retention of a farnesyl group (a carbonbased organic compound) to prelamin A, the precursor to lamin A. As stated above, the LMNA mutation associated with HGPS leads to the deletion of amino acids, including the amino acid which usually signals the cleavage site for the farnesyl group from prelamin A in order to form mature lamin A. Instead, the farnesyl group remains attached making progerin toxic to the nucleus. The team’s research suggests that this impacts the lamin A-retinoblastoma protein (pRB) signalling network, causing pRB to interact differently with its downstream transcription factors and regulators, thus implicating this network as a key factor in HGPS pathogenesis. Now that the mechanism starts to unravel, the team treated HGPS fibroblasts with a protein farnesyltransferase inhibitor (FTI) and found that this reversed abnormal gene expression in 288 of the 352 genes found in fibroblasts from HGPS patients. This outcome suggests that modulation of the lamin A-pRB signalling network could be a key factor in slowing down premature ageing.
A 2012 study by Professor Djabali and her colleagues provided the first in vivo evidence (evidence from within a living organism) that progerin is produced in adult stem cells (visualised on skin sections). Through the development of a new method of isolating naïve multipotent skin derived precursor cells from human fibroblast cultures, the team were able to observe a number of cell properties. These precursor cells had similar stem cell properties to cells isolated from skin biopsies. The cells could self-renew and differentiate in smooth muscle cells and fibroblasts. They were also found to express nestin (a marker of neuronal stem cells). A subset of these cells exhibited progerin positive signals which could reiterate the same molecular process which leads to progerin accumulation in adult stem cells. This study provides a viable alternative to skin biopsy samples from HGPS patients, which are not readily available for research, and opens new avenues to study the pathogenesis of other rare diseases.
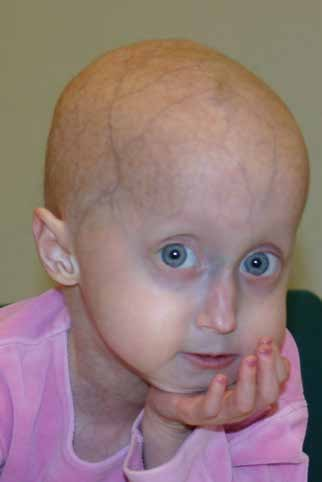
HGPS patient, image courtesy of PRF
In a 2014 study, Professor Djabali and her team investigated the use of sulforaphane (SFN) as a treatment for HGPS. SFN is an organic compound which can be found in cruciferous vegetables, such as young broccoli and cauliflower sprouts. The team aimed to test the ability of SFN to induce progerin clearance and improve disease phenotype in HGPS cell cultures using two-dimensional differential in a gel electrophoresis (2D-DIGE) approach. Progerin accumulation results in alterations to several components of protein degradation pathways. It leads to the impairment of autophagy (the cell’s ability to degrade and recycle cellular components) and proteasome activity (the breakdown of damaged or unneeded proteins). As a potent inducer of antioxidant and detoxification enzymes, SFN also enhances the protein degradation systems and promotes progerin clearance and proteostasis (the process by which cells control the quantity and folding of proteins). SFN does this through modulation of the nuclear factor erythroid 2-related factor (Nrf2) signalling pathway.
The Nrf2 signalling pathway can be looked upon as a ‘master regulator’ of antioxidant defence within the cells. Nrf2 targets genes which promote resistance to cancer and other diseases through the transcription of detoxifying and anti-inflammatory proteins. It may also play a role in preventing premature ageing. So how does SFN fit in? It’s all down to the interaction between SFN and a sensor known as Keap1. Keap1 is part of the body’s stress response system and inhibits Nrf2 signalling. SFN disrupts this process and allows Nrf2 to accumulate in the nucleus and activate the transcription of its target genes some of which were identified as downregulated in HGPS cells in the 2D-DIGE analysis.
In the study, cells treated with SFN exhibited significant increases in growth rate, proteasome activity and autophagy. SFN treatment also led to more efficient DNA damage repair in the cell, increased ATP levels and normalisation of the nuclear envelope. Significantly, the increase in proteostasis and progerin clearance remained high over 12 weeks, supporting its efficacy as a long term therapeutic strategy in cell based system and show potential for children with HGPS. Therefore, Professor Djabali’s team are conducting further studies to determine the extent to which modulation of the Nrf2 pathway with SFN could be a candidate target for future clinical trials.
Additionally, stimulating the Nrf2 pathway may be beneficial to normal cells, progerin and prelamin A also accumulate in some cells of the skin and vascular system in unaffected individuals. Because the number of cells expressing this abnormal protein – progerin – seems to increase with age, further studies are underway to test the potential of manipulating this pathway in the context of physiological ageing. Remarkably, these studies show once again that important insight into the ageing process can be gained by studying HGPS. HGPS may not only pinpoint therapeutic avenues for fighting this rare genetic disorder, but might also teach us more about normal ageing and provide clues for how to slowdown ageing processes in general.
More recently, the findings of a 2016 study by Professor Djabali and her colleagues further explain the underlying mechanisms of genomic instability in HGPS cells. By investigating the effects of progerin on the dynamics of mitosis, the team uncovered evidence which appears to directly link progerin action to defects in chromosome segregation, nuclear envelope reformation and other cell defects. The first part of the study analysed the distribution of proteins in relation to the components of the nuclear lamina and envelope during mitosis. The findings showed that progerin began to cause defects in chromosome segregation as early as the metaphase (shortly after the breakdown of the nuclear envelope). This led to delays in nuclear envelope reformation as well as trapped cell components and proteins in the endoplasmic reticulum during cell division.
The second part of the investigation looked at the effect of progerin on the localisation of two proteins, CENP-E and CENP-F, on kinetochores. Kinetochores are protein structures which allow chromatids to attach to spindle fibres and are required for proper segregation of chromosomes. While distribution of CENP-E was similar to that in normal cells, CENP-F was displaced from kinetochores during the metaphase. This suggests that CENP-F could be a target of progerin during mitosis. The team also found evidence of increased chromatin lagging and genomic instability which increased proportionally with progerin levels. These progerin dependent defects accumulate with each round of mitosis, suggesting that high levels of progerin predispose cells to undergo premature senescence.
Delaying Age Related Disease
There is also a lot to learn from this research regarding physiological ageing outside of disease processes. By examining the processes underlying premature ageing syndromes, Professor Djabali and her colleagues can determine whether or not similar mechanisms underlie normal ageing. It may also lead to insights in the pathology underlying other age related diseases, particularly vascular and neurological disorders affecting the elderly population. Therefore, the team aim to define isolate and characterise skin stem cells in order to determine how ageing might impact stem cell renewal and differentiation. This unique approach to the study of ageing makes the laboratory the only one in Germany to examine the characteristics of ageing from this angle.
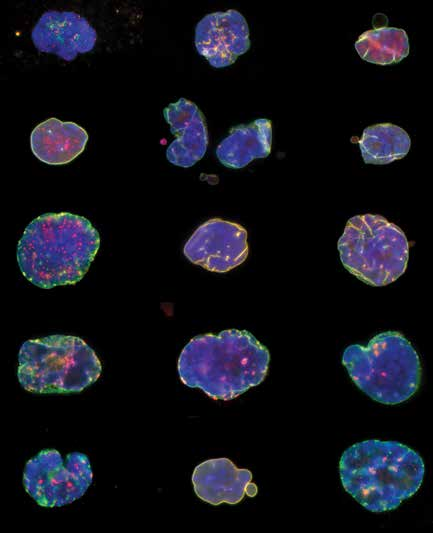
Collage of different nuclei from HGPS cells – all deformed instead of being ovoid in shape
So far, studies into HGPS have led to some interesting discoveries in the processes of ageing. An examination of skin biopsies from individuals across the life span showed a comparable frequency of progerin at a low level in all age groups, but increased levels and deeper distribution of the protein were found in the older population. This suggests that progerin expression could be used as a biomarker for normal cellular ageing. Likewise, in a histological comparison of vascular pathology in HGPS and vascular pathology of ageing, the team and collaborators detected progerin in both samples, supporting the theory that progerin has a role in cardiovascular ageing amongst the general population. Similar results were found in a later study examining the link between atherosclerosis in ancient humans, premature ageing syndromes and normal ageing. After imaging studies of ancient mummies revealed vascular calcification, researchers became interested in the possibility of a genetic predisposition to atherosclerosis. In their 2014 study, the team found that progerin can be detected in vascular cells. The resulting loss in cells from the vascular walls and replacement by fibrous tissues promotes hardening and calcification of the blood vessels. Although vascular progerin is found in low levels in the general population, these levels increase with age, suggesting a potential role for progerin in age-related atherosclerotic processes.
Next steps in ageing research
So what’s next for Professor Djabali and her team? As well as continuing her search for a cure for HGPS, the knowledge gained from the research on progeria could drive future research in Alzheimer’s disease (AD). By dissecting the underlying mechanisms of ageing cells in patients with AD, it may be possible to better understand which processes are inducing neuronal degeneration.
Research into treatments for HGPS may also be useful in identifying new targets for cancer therapy. In simple terms, cancer is the uncontrolled division and growth of cells. One of the major risk factors for cancer is ageing. What is remarkably intriguing is that cellular ageing leads to cell cycle exit via down regulating signalling pathways controlling the proliferation of cells. In cancer some of the overlapping signalling triggers the opposite outcome. How does the balance between these signals dictate such opposite cellular fate? Can HGPS teach us which signalling or gene targets could lead to anti-cancer treatments? Professor Djabali is searching for answers.
Finally, a greater understanding of how adult stem cells work can inform work in regenerative medicine and drug therapies. Studying the mechanisms by which protein breakdown regulates the processes of ageing could be the key to identifying therapeutic interventions which promote healthy cellular function and ameliorate age-related pathologies.
Meet the researcher

Professor Karima Djabali
Epigenetics of Aging
TUM School of Medicine
Department of Dermatology
Technical University Munich (TUM)
Germany
E: djabali@tum.de
T: (+49) 89 289 10920
W: www.professoren.tum.de/en/djabali-karima
V Eisch, X Lu, D Gabriel and K Djabali, Progerin impairs chromosome maintenance by depleting CENP-F from metaphase kinetochores in Hutchinson-Gilford progeria fibroblasts, Oncotarget, 2016. DOI: 10.18632/oncotarget.8267
D Gabriel, D Roedl, LB Gordon and K Djabali, Sulforaphane enhances progerin clearance in Hutchinson–Gilford progeria fibroblasts, Aging Cell, 2015, 14, 78–91.
V Wenzel, D Roedl, D Gabriel, LB Gordon, M Herlyn, R Schneider, J Ring and K Djabali, Naive adult stem cells from patients with Hutchinson- Gilford progeria syndrome express low levels of progerin in vivo, Biology Open, 2012, 1, 516–526.
J Marji, SI O’Donoghue, D McClintock, VP Satagopam, R Schneider, D Ratner, HJ Worman, LB Gordon, K Djabali, Defective Lamin A-Rb Signaling in Hutchinson-Gilford Progeria Syndrome and Reversal by Farnesyltransferase Inhibition, PLoS ONE, 2010, e11132.
D McClintock, D Ratner, M Lokuge, DM Owens, LB Gordon, FS Collins and K Djabali, The mutant form of lamin A that causes Hutchinson- Gilford progeria is a biomarker of cellular aging in human skin, PLoS One, 2007, 2, e1269.
D McClintock, LB Gordon and K Djabali, Hutchinson-Gilford progeria mutant lamin A primarily targets human vascular cells as detected by an anti-Lamin A G608G antibody, Proc Natl Acad Sci USA, 2006, 103, 2154–9.

Download
{kind=link}