
Arthur Schwartz | Mechanistic Insights into the Therapeutic Potential of Dehydroepiandrosterone Analogues
Dehydroepiandrosterone (DHEA) is the most abundant steroid hormone in the bloodstream, although it declines significantly with age. DHEA therapeutics could have a role in the development of anti-ageing preventative medicine. Professor Emeritus Arthur Schwartz of Temple University and his colleagues at Sterotherapeutics LLC, are developing a DHEA analogue, fluasterone, which is far more potent than native DHEA and lacks DHEA’s androgenic and estrogenic side effects. Professor Schwartz is working to explain the mechanistic underpinnings of DHEA’s health benefits.
DHEA – The Enigmatic Sixth Steroid Hormone
The human endocrine system is based on six main steroid hormones. You have probably heard of the sex hormones estradiol, progesterone and testosterone, cortisol, the ‘stress hormone’ and aldosterone, the ‘blood pressure hormone’. The sixth hormone, dehydroepiandrosterone (DHEA), is much less widely known or understood.
DHEA, as a sulphated ester, DHEA(S), is the most abundant steroid in the bloodstream. Our plasma DHEA(S) levels peak in our twenties but then decline with age. By the time we reach our eighties and nineties, our DHEA levels are 5–10% of what they were in our twenties. Professor Arthur Schwartz of Temple University believes DHEA could have a place in anti-ageing preventative medicine. Epidemiological studies have shown that DHEA(S) deficiency is related to poorer physical fitness, increased cardiovascular, and critically, all-cause mortality risk. Animal studies have shown that DHEA supplementation can mitigate against cancer, atherosclerosis, obesity, and diabetes. Can this translate to humans?
Initial clinical findings have been underwhelming. In a clinical trial conducted in 2006, older adults received daily oral DHEA supplements at doses 50 or 75 mg in women and men, respectively. Disappointingly, no beneficial health effects were apparent. However, Professor Schwartz remains optimistic, noting that in this trial, dosages were selected so that serum DHEA(S) levels would increase to the peak levels of people in their twenties. If extrapolated to humans, DHEA dosages that elicit health benefits in animal models would be 15 to 30 times the 75 mg dosage used in the human trial and could elicit untoward androgenic and estrogenic side effects. Therefore, a more potent analogue lacking the sex-hormonal side effects of DHEA is needed.
Professor Schwartz is a scientific advisor to Sterotherapeutics LLC, a pharmaceutical start-up company. Professor Schwartz and his colleagues have synthesised and screened over 50 synthetic and natural analogues of DHEA in mouse models. One analogue, 16α-fluoro-5-androsten-17-one, named fluasterone, stood out as far more potent than DHEA. In mouse studies, Professor Schwartz showed that fluasterone has superior cancer preventive, anti-glucocorticoid, anti-diabetic, and anti-obesity properties compared with DHEA. Furthermore, unlike native DHEA, fluasterone is non-androgenic and non-estrogenic, so it should avoid these adverse effects. Using mouse models, he delves into the mechanistic underpinnings of DHEA’s mode of action – these largely revolve around inhibiting the release of reactive oxygen species (ROS) and antagonising glucocorticoid (GC) activity.
Anti-Cancer Effects
ROS are oxygen-derived radicals that are highly reactive oxidising agents. They have the potential to wreak immense oxidative damage on biological molecules and cells. Fortunately, cells have various mechanisms to avoid excessive ROS generation – DHEA forms part of this. ROS are generated by various enzymes, including cytochrome p450 and NADPH oxidases (NOX). NOX enzymes are widely distributed and produce ROS as their main product. Although the biological roles of NOX enzymes are still being worked out, phagocytes are known to deploy NOX 2 to release bursts of ROS to fight microbial pathogens. However, excessive NOX activity is thought to be a hidden driver in age-related diseases, including cancer, atherosclerosis, ischemic stroke, fibrosis, and neurodegeneration.
The pentose phosphate pathway (PPP) generates pentoses (five-carbon sugars), which are nucleotide precursors. Glucose-6-phosphate (G6P) is an important metabolite, forking into either glycolysis or the PPP. In the initial phase of the PPP, G6P is converted to 6-phosphogluconate, catalysed by glucose-6-phosphate dehydrogenase (G6PD). This conversion leads to NADP+ being reduced to NADPH. NADPH is critical for ROS generation, being used as a reducing agent by oxidative enzymes, including NOX and cytochrome p450. DHEA is a potent uncompetitive inhibitor of G6PD and, therefore, prevents the generation of NADPH, starving oxidative enzymes of this vital substrate.
In the well-studied two-stage skin tumorigenesis model in mice, skin papillomas are induced by topically applying a carcinogen such as 7,12-dimethylbenz (a)anthracene (DMBA), followed by twice weekly applications of a tumour promoter, such as tetradecanoylphorbol-13-acetate (TPA). DMBA is converted to an active carcinogen-mutagen by an NADPH-dependent cytochrome p450. Repeated TPA application produces sustained inflammation and epidermal hyperplasia, which acts on the DMBA-mutated cells to promote papilloma formation. Treating mice with DHEA inhibits papilloma formation at both the DMBA-initiation and TPA-promotion stages.
In Professor Schwartz’s studies, fluasterone was more potent than DHEA in blocking DMBA activation as well as TPA-induced inflammation, hyperplasia, and papilloma formation. Notably, fluasterone’s anti-cancer effect was reversed by supplying the mice with drinking water containing the four deoxyribonucleosides, which overcomes the G6PD-induced inhibition in five-carbon sugars as well as very likely increases NADPH levels. This suggests that G6PD inhibition is critical to fluasterone’s anti-tumour activity.
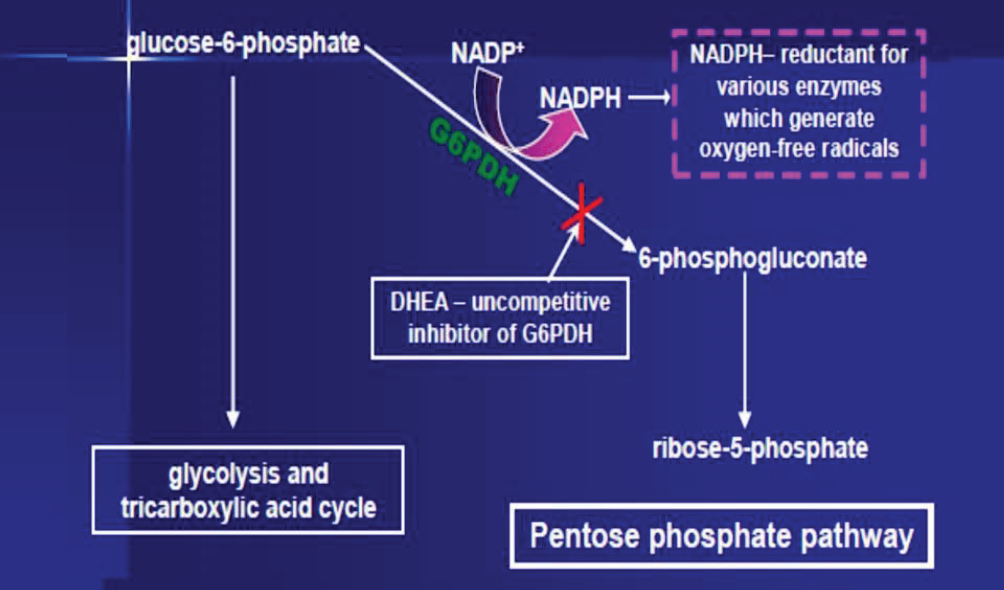
The biochemical pathway inhibited by dehydroepiandrosterone. Reproduced with permission from Arthur G. Schwartz under the terms of a Creative Commons attribution license. DOI: https://doi.org/10.14336/AD.2021.0913 Credits: Arthur G Schwartz
Antagonising Glucocorticoid Activity
Cortisol is a GC hormone that primes the body for stressful situations – but too much cortisol is definitely problematic. Prolonged elevated cortisol exposure is a feature of Cushing’s syndrome – this may be due to excessive secretion (endogenous) or overuse of GC medication (exogenous). Serum cortisol increases with age – in tandem with the decline in DHEA. Professor Schwartz hypothesises that this interplay could be causally associated with age-related morbidities. These morbidities – experienced by patients with Cushing’s syndrome – include weight gain, hyperglycaemia, hypertension, immunosuppression, as well as osteoporosis and muscle atrophy.
Preclinical studies have shown DHEA to be a GC antagonist. In mouse and rat models exposed to GC drugs, negative effects were mitigated with DHEA. Dexamethasone (Dex) is a GC drug with about 40 times the potency of cortisol. The effects of Dex exposure in mouse models include shrinking of the thymus and spleen (thymic and splenic atrophy), suppression of T- and B-lymphocytes, hypertension, and susceptibility to viral infection. In previous studies, these Dex-associated effects were antagonised in mice treated with DHEA.
Professor Schwartz’s team compared the GC-antagonistic effect of eight DHEA analogues in mice exposed to Dex. They found that the analogues protected the mice against thymic and splenic atrophy and weight gain. Fluasterone was the most potent analogue, six times more potent than native DHEA in protecting against atrophy. Notably, they found a correlation between the analogues’ anti-GC and anti-obesity activity, consistent with the known weight-enhancing effect of GC.
Mechanistic Speculations
How does DHEA antagonise GC activity? DHEA does not bind to the GC receptor and is not a competitive inhibitor. Preclinical studies have provided some intriguing mechanistic clues. In vitro cell culture and in vivo mouse studies suggest that DHEA reduces the activity of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1), an enzyme that ‘reactivates’ GC. Both preclinical and clinical studies have shown that reducing the activity of 11β-HSD1 confers resistance to excess cortisol exposure. However, Professor Schwartz thinks that inhibition of 11β-HSD1 may partly account for DHEA’s anti-GC action but is unlikely to explain its protection against high-dose Dex treatment in animal studies. Other mechanisms must play a role.
A Tale of Two Receptors
Two oestrogen receptors – alpha and beta (ERα and ERβ) – are important in cancer. Activation of ERα, the classical oestrogen receptor, can promote breast and uterine cancer, whereas activation of ERβ antagonises ERα activity. Importantly, numerous preclinical studies have demonstrated that ligand-activated ERβ acts as a broad-spectrum tumour suppressor. Many studies have shown that DHEA, as well as a specific DHEA metabolite, 5-androstene-3β,17β-diol (ADIOL), bind to and transcriptionally activate ERβ. ADIOL is significantly more active than DHEA and is believed to function as a potent endogenous activator of ERβ. Professor Schwartz suggests that DHEA-fluasterone suppresses cancer development in animal studies through ERβ activation as well as through G6PD inhibition.
The Move into Clinical Development
Preclinical mouse models are shedding new light on the disease-ameliorating effects of fluasterone. Professor Schwartz now wants to take this work into clinical development. Fluasterone will shortly enter a phase 2 trial for treating hyperglycaemia in patients with endogenous Cushing’s syndrome. If successful, this would validate the anti-glucocorticoid action of fluasterone in humans and pave the way for additional translational studies to assess the therapeutic and preventive efficacy of fluasterone.

SHARE
{kind=link}
DOWNLOAD E-BOOK

REFERENCE
https://doi.org/10.33548/SCIENTIA992
MEET THE RESEARCHER

Professor Arthur G Schwartz
Sterotherapeutics LLC
Doylestown, PA
USA
Arthur Schwartz, Professor Emeritus of Microbiology at Temple University’s Health Sciences Center, is one of the world’s leading researchers on the steroid hormone dehydroepiandrosterone (DHEA), having spent over four decades investigating its chemical, biochemical, and physiological properties. He obtained his BA at Johns Hopkins University in 1961 and his PhD at Harvard University in 1968. He then embarked on postdoctoral fellowships at Oxford University and the Albert Einstein College of Medicine, New York, before taking on an assistant professorship at Temple University in 1972. He remains at Temple, having risen through the academic ranks. He is interested in applying DHEA analogues to prevent and treat age-related diseases, particularly cancer chemoprevention. To this end, he is a Scientific Advisor for SteroTherapeutics LLC, a pharmaceutical start-up company he helped establish in 2016. Professor Schwartz is an avid inventor, having obtained over 14 patents, most of which relate to DHEA analogue therapy.
CONTACT
KEY COLLABORATORS
Dr Laura L Pashko, Temple University
Dr Marvin L Lewbart, Steroid Laboratory, Voorhees, NJ (deceased)
Dr John R Williams, Temple University
Dr G Chris Christensen, Jefferson-Abington Hospital
Dr Manohar Katakam, Sterotherapeutics
Dr Constantine Stratakis, Sterotherapeutics
Dr Bo Allen, Sterotherapeutics
FUNDING
National Cancer Institute
National Institute on Aging
American Cancer Society
American Institute for Cancer Research
Advanced Technology Center of Southeastern Pennsylvania
Samuel S. Fels Fund
Research Corporation Technologies
FURTHER READING
AG Schwartz, Dehydroepiandrosterone, Cancer, and Aging, Aging and Disease, 2022, 13(2), 423–432. DOI: https://doi.org/10.14336/AD.2021.0913

REPUBLISH OUR ARTICLES
We encourage all formats of sharing and republishing of our articles. Whether you want to host on your website, publication or blog, we welcome this. Find out more
Creative Commons Licence (CC BY 4.0)
This work is licensed under a Creative Commons Attribution 4.0 International License. 
What does this mean?
Share: You can copy and redistribute the material in any medium or format
Adapt: You can change, and build upon the material for any purpose, even commercially.
Credit: You must give appropriate credit, provide a link to the license, and indicate if changes were made.
SUBSCRIBE NOW
Follow Us
MORE ARTICLES YOU MAY LIKE

Assoc Prof. Nicholas Brown | Rethinking Prostate Care: A New Frontier in Treating Benign Prostatic Hyperplasia
For millions of men, ageing brings with it a set of frustrating and often disruptive urinary symptoms. These symptoms, caused by benign prostatic hyperplasia, or BPH, can affect sleep, confidence, and overall quality of life. Traditionally, treatment follows a familiar path. Patients begin with medications, often for years, and may eventually progress to surgery if symptoms worsen. Yet this pathway is not without its drawbacks. Medications can cause side effects, while surgery carries risks and recovery time. In recent years, a minimally invasive interventional radiology procedure called prostate artery embolisation, or PAE, has begun to challenge this traditional model. At the forefront of this shift is a collaborative research group, led by Dr. Nicholas Brown of the University of Queensland, whose series of P-EASY studies has explored whether PAE could transform how BPH is treated, particularly at earlier stages.

Jean Lycke | Addressing Unmet Medical Needs in Mucosal Disease: A Close-to-Market Innovation Approach
Recurrent Aphthous Stomatitis (RAS) is an oral condition characterized by one or several painful mucosal ulcers. RAS affects a large proportion of the population and has a point prevalence of approximately 2–3%, daily. The etiology remains unknown, and there is currently no curative treatment. Most patients experience recurring episodes over time, with each episode typically lasting up to a week. Here, we describe the development of a mucoadhesive patch which, when applied over a RAS ulcer, provides rapid pain relief. The patch is easy for patients to apply when symptoms begin and has the potential to be used as an over-the-counter product. The development of the Mucocort mucoadhesive patch is an example of a Close-to-Market innovation strategy that embraces simplicity within a complex healthcare system. By simplifying the product concept, the team has reduced the number of regulatory steps required before market approval. This MedTech/Pharma innovation model, known as the “4R” framework – Re-purposing, Re-formulation, Re-positioning, and Re-patenting – has guided the program from concept to commercialization. In addition to the biodegradable mucoadhesive patch developed for RAS ulcers, the team is extending the innovation concept to a mucoadhesive gel formulation for the prevention and treatment of chemotherapy-induced mucositis. This gel-based program is being commercialized separately through MucoShield.
The Translational Asian Agerelated Macular Degeneration Program Phase 2 (TAAP-2): Reimagining the Future of Vision Care
Age-related macular degeneration, often abbreviated as AMD, is one of the leading causes of vision loss among older adults worldwide. In Asia, where populations are ageing rapidly, its impact is particularly profound. For many, the disease quietly erodes central vision, making everyday activities such as reading, driving, and recognising faces increasingly difficult. Against this backdrop, the Translational Asian Age-related Macular Degeneration Programme, or TAAP for short, has emerged as a bold and ambitious effort to confront the disease headon. Now in its second phase, TAAP-2 represents a significant evolution in both scientific scope and clinical ambition.

Ms. Aikaterini Dritsoula | Looking Beyond Snoring: How Hidden Airway Problems Shape Children’s Sleep
For many parents, a child’s snoring may seem harmless, even endearing. Yet in some cases, it signals something more serious. Obstructive sleep apnoea is a condition in which a child’s breathing is repeatedly disrupted during sleep. These interruptions can affect growth, behaviour, and learning. Children with this condition may toss and turn at night, struggle to concentrate during the day, or show signs of hyperactivity and fatigue. Traditionally, enlarged tonsils and adenoids have been seen as the main culprits. Surgery to remove them has long been considered the standard treatment. However, research led by Consultant ENT Surgeon Ms. Aikaterini Dritsoula of The Leeds Teaching Hospitals NHS Trust invites us to look deeper. Her work suggests that the story is often more complex, especially in very young children.