
Dr. David Westaway – Misfolding of Brain Proteins Triggering Neurodegenerative Diseases
Our DNA codes for proteins that are essential for the normal structure and function of our cells, tissues and organs. These proteins are folded in specific ways to facilitate these functions, but in disease states, this folding can go wrong. Dr. David Westaway from the University of Alberta in Canada investigates how and why protein misfolding occurs and how strains of misfolded proteins result in neurodegenerative diseases like dementia. His research is paving the way for novel therapies for these currently incurable and devasting conditions.
The Structure and Function of Proteins in Our Cells
Proteins are large and complex molecules that are found in every cell of the human body. They have an extensive number of different roles, ranging from making up structures within cells to carrying out specific functional roles or regulating other processes for normal body tissue and organ function. One example of a structural protein is called actin; the actin protein gives structure and support to cells and allows proper body movement. Antibodies that protect our bodies from infection are functional proteins, whereas enzymes are regulatory proteins that that help to carry out vital chemical reactions such as turning food into energy.
Each different protein is coded for by specific sequences of DNA, known as genes. Once cell machinery has deciphered these genes and determined which proteins are required, it joins together the relevant amino acids in the correct order to make these proteins. There are 20 different amino acids that can be stitched together in different orders and amounts to create a vast number of protein possibilities. This order of amino acids is known as a protein’s primary structure.
For proteins to function correctly, the secondary and tertiary structures, as well as the primary structure of the protein must be exact. The secondary structure is the local interactions between smaller sections of the protein whilst the tertiary structure is the overall, three-dimensional folding and structure of the protein. These three-dimensional shapes and their associated rigidity – or sometimes, aspects of their flexibility – are what allow a protein to perform its duties and keep cells healthy.

What Happens When Protein Folding Goes Wrong in the Brain?
Unfortunately, protein folding does not always go according to plan. In neurodegenerative brain diseases, a small group of proteins can be incorrectly folded and then cluster together. These clusters of proteins become sufficiently large that they can easily be seen with a simple microscope, and are observed to lie inside or next to brain cells.
To make matters worse, once these clusters have started to form, they can catalyse their own formation by taking new, normally folded proteins and disrupting their structure to be misfolded. In this process, the normal proteins are known as substrates and the misfolded proteins are collectively known as products.
Often, the result of a catalytic misfolding process is a neurodegenerative disease whereby brain function continues to decline over time. This loss of brain function can be associated with several different symptoms including memory loss, difficulty of movement, trouble speaking and unpredictable moods. Profound loss of reasoning and memory function is synonymous with the umbrella term dementia. One of the most common types of dementia is Alzheimer’s disease, which is well-known to affect a brain structure called the hippocampus while frontotemporal dementia produces degeneration nearer to the front of the brain. Apart from rare cases where substrate proteins are predisposed to misfold by a mutation, dementias are associated with older age and there is evidence that different lifestyle factors can either advance or stave off their development.
Unfortunately, around one-sixth of people over the age of 80 have dementia and the World Health Organization estimates 50 million people live with it worldwide. Whilst there are some medications available to ease the symptoms of dementia, the first generation of curatives is proving controversial and so, where possible, alternative treatments may be suggested. This might include cognitive stimulation therapy to improve brain function or cognitive rehabilitation to facilitate everyday tasks. Nevertheless, due to erosion of memory, intellect and normal behaviours, neurodegenerative diseases remain a difficult issue for many people and their families. Understanding the underlying events through research is an important step towards progressing different mechanism-based therapeutics.
Studying the Molecular Origins of Neurodegenerative Diseases
Unravelling the reasons for, mechanisms behind and solutions to neurodegenerative disease processes is Dr. David Westaway from the Centre for Prions and Protein Folding Diseases at the University of Alberta in Canada. He describes how the series of steps leading to the assembly of misfolded proteins is complex and the outcomes for any given misfolded protein are diverse. As a result, different types of protein clusters can form in the brain called ‘strains’ – and superficially similar to viral strains we are now all familiar with because they have certain predictable properties. Different misfolded protein strains can react differently to medications too; whereas one strain may diminish and respond well to a certain treatment in petri-dish experiments, another strain may partially or completely evade treatment.
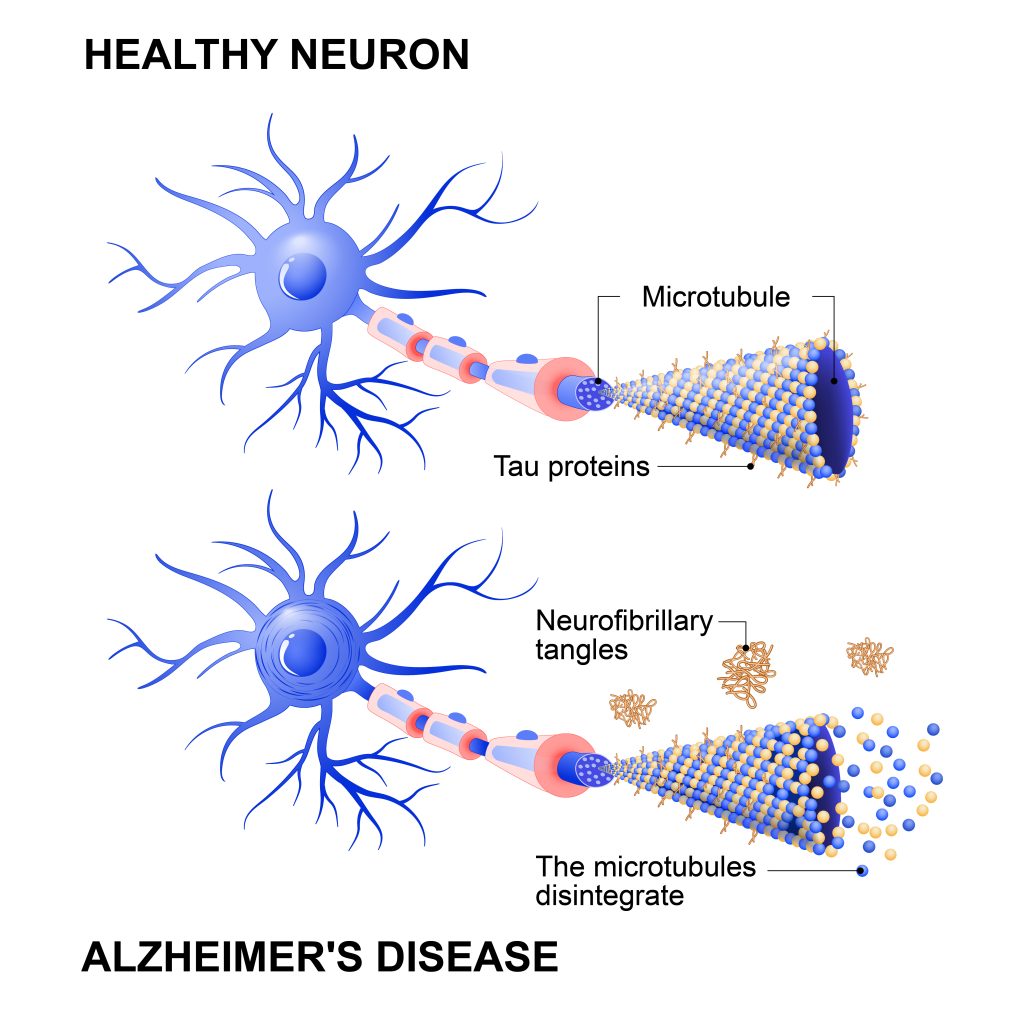
According to Dr. Westaway, ‘My laboratory focuses on these issues and events for two proteins of interest – the cellular prion protein and the protein tau’. The cellular prion protein is found on the surface of cells in many different tissues and organs but especially in the central and peripheral nervous systems. As the brain is a part of the central nervous system, when the cellular surface protein becomes misfolded and builds up it causes neurodegenerative diseases. These are collectively referred to as prion diseases. Tau proteins are also found in abundance in the central nervous system and neurons, and abnormal accumulations of tau are associated with Alzheimer’s and Pick’s disease, which are also neurodegenerative syndromes. The collective name for tau diseases is tauopathies.

Misfolding of the Cellular Prion Protein
Before scientists were aware of different strains of neurodegenerative disease proteins, it was generally believed that one type of amino acid chain could only result in one type of three-dimensional folding and one type of misfolding. This perspective has now been superseded and Dr. Westaway explains that there is ‘another layer of complexity – the misfolding outcomes for a protein can be diverse and can change depending on which part of the molecule is being considered.’
This has previously been studied and documented in a protein called amyloid precursor protein in relation to Alzheimer’s disease. Following on from this, Dr. Westaway and his team have looked into cellular prion protein and how it is involved in disease development. They have studied mutant types of the cellular prion protein that are predisposed to misfolding and inevitably cause disease.
A mutant protein has single or multiple amino acids replacing those that are standard, resulting in an abnormal protein. This is due to genes that are themselves coded abnormally or alternatively, it is due to a mistake in protein quality control somewhere along the production line in cells. Dr. Westaway is interested in finding and developing chemicals and drugs that could control misfolding events due to mutant proteins. This could lead to exciting developments in therapeutics for multiple neurodegenerative diseases.

Strains of the Tau Protein Lead to Different types of Dementias
Dr. Westaway has also moved forward our understanding of changes in tau in relation to different types of tauopathy. Describing his work, Dr. Westaway notes, ‘for the protein tau, using susceptible mice made by genetic engineering and a chemical test of misfolding, we have shown that one type of substrate molecule can create a collection of different misfolded variants; this work was carried out with our collaborator Jiri G. Safar, MD. As elapsed time increased beyond two-thirds of the natural lifespan of these mice, this mixture of variants evolved into two or three clearly recognisable strains of misfolded tau protein.’
This means that he was able to demonstrate one protein misfolding into multiple strains in a real-world example. In animal models of disease (e.g., mice), disease progression is sometimes deliberately accelerated to make experiments quicker, not to mention less expensive. However, this can mean that the processes that result in different tau strains are difficult to detect. Therefore, Dr. Westaway utilised a ‘slow’ model of neurodegenerative disease and this allowed him to study the evolution of events that lead up to strain formation.
The team found that, in a human context that shares the same type of tau protein as the mouse disease model, the different tau protein strains were associated with different types of dementia that affect the frontal lobes of the brain (collectively known as frontotemporal dementias). Here, the suspicion was – and has now been verified by chemical analyses of human and animal model samples – that the differences that could be seen in patients behaviours originated from slightly different forms of abnormal tau.
‘My laboratory is tracking down the chemical processes and cells which participate in the evolution of toxic misfolded forms of the tau protein’, says Dr. Westaway. If these processes can be thoroughly defined, they can be targeted by novel therapies for the disease. Blocking the formation and diversification of these unwanted strains through drugs could be a promising method of preventing and blocking neurodegenerative diseases.
Continuing the Vital Work: New Funding
Recently, Dr. Westaway and his colleagues in the Centre for Prions and Protein Folding Diseases, in the Neuroscience and Mental Health Institute and in the Department of Physics at the University of Alberta, have secured novel federal, provincial and for-profit funding of an incredible $9.5 million. This backing came thanks to an investment initiative of over half a billion dollars led by Canada’s Prime Minister, Justin Trudeau. The goal of distributing this funding is to sustain innovative, scientific research and discovery within the country. This exciting financial support will help Dr. Westaway and his colleagues carry on advanced research into neurodegenerative diseases.
Already, Dr. Westaway and his team have made impressive strides into deepening our understanding of protein folding and misfolding and how they impact health and disease. By elucidating the processes that result in protein strains and consequent neurodegenerative disease, he is beginning to point towards possible new therapies for the future. This provides important hope for the millions of people suffering from dementia worldwide.
SHARE
{kind=link}
DOWNLOAD E-BOOK

REFERENCE
https://doi.org/10.33548/SCIENTIA766
MEET THE RESEARCHER

Dr. David Westaway
Centre for Prions and Protein Folding Diseases
University of Alberta
Edmonton, Alberta
Canada
Dr. David Westaway completed his BSc in Biochemistry at the University of Sussex in the UK, and went on to achieve a PhD in the Biochemistry Department of Imperial College at the University of London. Having received multiple awards in his field and filled numerous academic positions, Dr. Westaway is now a Professor in the Department of Medicine (Neurology) at the University of Alberta in Canada. He is also the Director of the Centre for Prions and Protein Folding Disease at the University of Alberta, which is where he carries out his research into what happens when protein folding goes wrong in our cells.
CONTACT
E: david.westaway@ualberta.ca
FURTHER READING
N Daude, C Kim, S-G Kang, et al., Diverse, evolving conformer populations drive distinct phenotypes in frontotemporal lobar degeneration caused by the same MAPT‑P301L mutation, Acta Neuropathologica, 2020, 139(6), 1045–1070.
AR Castle, N Daude, S Gilch, D Westaway, Application of high-throughput, capillary-based Western analysis to modulated cleavage of the cellular prion protein, Journal of Biological Chemistry, 2019, 294(8), 2642–5291.
A Lau, A McDonald, N Daude, et al., Octarepeat region flexibility impacts prion function, endoproteolysis and disease manifestation, EMBO Molecular Medicine, 2015, 7(3), 339–356.
![]()
REPUBLISH OUR ARTICLES
We encourage all formats of sharing and republishing of our articles. Whether you want to host on your website, publication or blog, we welcome this. Find out more
Creative Commons Licence (CC BY 4.0)
This work is licensed under a Creative Commons Attribution 4.0 International License. 
What does this mean?
Share: You can copy and redistribute the material in any medium or format
Adapt: You can change, and build upon the material for any purpose, even commercially.
Credit: You must give appropriate credit, provide a link to the license, and indicate if changes were made.
SUBSCRIBE NOW
Follow Us
MORE ARTICLES YOU MAY LIKE

Dr. Colleen Fisher | Singing to the Lions: Tackling Childhood Trauma in Zimbabwe Through Creative Psychosocial Support
Mental health challenges are one of the biggest issues facing children and adolescents across the world, particularly in low-resource settings. In Zimbabwe, a 3-day intervention called Singing to the Lions is helping young people build resilience through creative, traumainformed workshops. Dr Colleen Fisher and colleagues in Zimbabwe and the U.S. led the first formal evaluation of the intervention, offering new insight into how locally grounded intervention approaches can support youth mental health at scale.

Assoc Prof. Nicholas Brown | Rethinking Prostate Care: A New Frontier in Treating Benign Prostatic Hyperplasia
For millions of men, ageing brings with it a set of frustrating and often disruptive urinary symptoms. These symptoms, caused by benign prostatic hyperplasia, or BPH, can affect sleep, confidence, and overall quality of life. Traditionally, treatment follows a familiar path. Patients begin with medications, often for years, and may eventually progress to surgery if symptoms worsen. Yet this pathway is not without its drawbacks. Medications can cause side effects, while surgery carries risks and recovery time. In recent years, a minimally invasive interventional radiology procedure called prostate artery embolisation, or PAE, has begun to challenge this traditional model. At the forefront of this shift is a collaborative research group, led by Dr. Nicholas Brown of the University of Queensland, whose series of P-EASY studies has explored whether PAE could transform how BPH is treated, particularly at earlier stages.

Jean Lycke | Addressing Unmet Medical Needs in Mucosal Disease: A Close-to-Market Innovation Approach
Recurrent Aphthous Stomatitis (RAS) is an oral condition characterized by one or several painful mucosal ulcers. RAS affects a large proportion of the population and has a point prevalence of approximately 2–3%, daily. The etiology remains unknown, and there is currently no curative treatment. Most patients experience recurring episodes over time, with each episode typically lasting up to a week. Here, we describe the development of a mucoadhesive patch which, when applied over a RAS ulcer, provides rapid pain relief. The patch is easy for patients to apply when symptoms begin and has the potential to be used as an over-the-counter product. The development of the Mucocort mucoadhesive patch is an example of a Close-to-Market innovation strategy that embraces simplicity within a complex healthcare system. By simplifying the product concept, the team has reduced the number of regulatory steps required before market approval. This MedTech/Pharma innovation model, known as the “4R” framework – Re-purposing, Re-formulation, Re-positioning, and Re-patenting – has guided the program from concept to commercialization. In addition to the biodegradable mucoadhesive patch developed for RAS ulcers, the team is extending the innovation concept to a mucoadhesive gel formulation for the prevention and treatment of chemotherapy-induced mucositis. This gel-based program is being commercialized separately through MucoShield.
The Translational Asian Agerelated Macular Degeneration Program Phase 2 (TAAP-2): Reimagining the Future of Vision Care
Age-related macular degeneration, often abbreviated as AMD, is one of the leading causes of vision loss among older adults worldwide. In Asia, where populations are ageing rapidly, its impact is particularly profound. For many, the disease quietly erodes central vision, making everyday activities such as reading, driving, and recognising faces increasingly difficult. Against this backdrop, the Translational Asian Age-related Macular Degeneration Programme, or TAAP for short, has emerged as a bold and ambitious effort to confront the disease headon. Now in its second phase, TAAP-2 represents a significant evolution in both scientific scope and clinical ambition.